

DNA damage during reoxygenation elicits a Chk2-dependent checkpoint response
- PMID: 16478982
- PMCID: PMC1430245
- DOI: 10.1128/MCB.26.5.1598-1609.2006
DNA damage during reoxygenation elicits a Chk2-dependent checkpoint response
Abstract
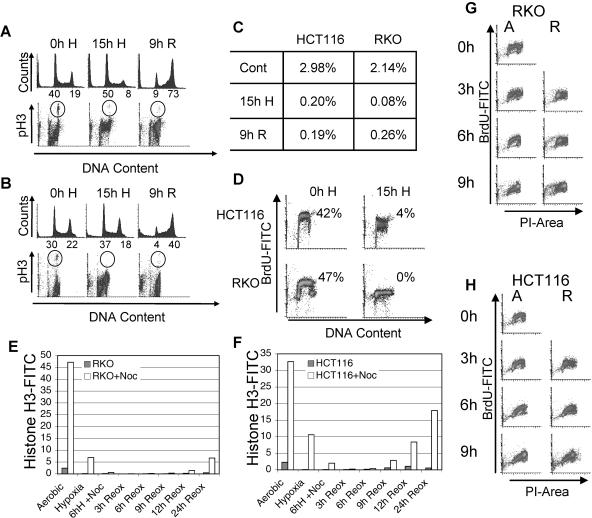
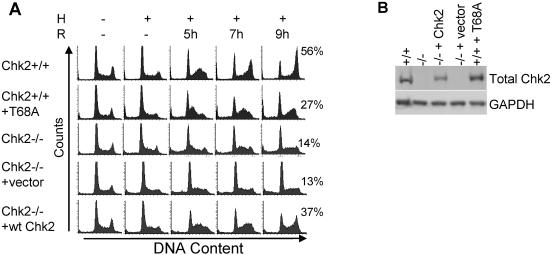
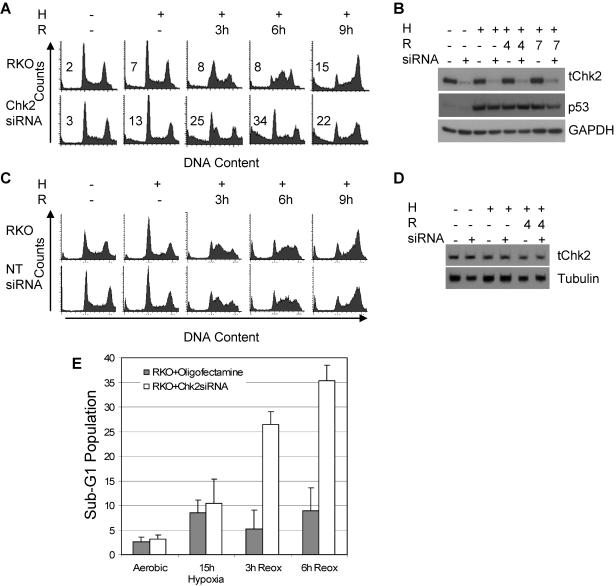
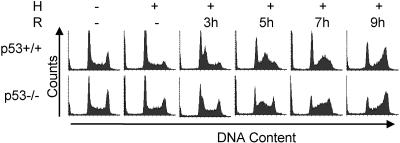
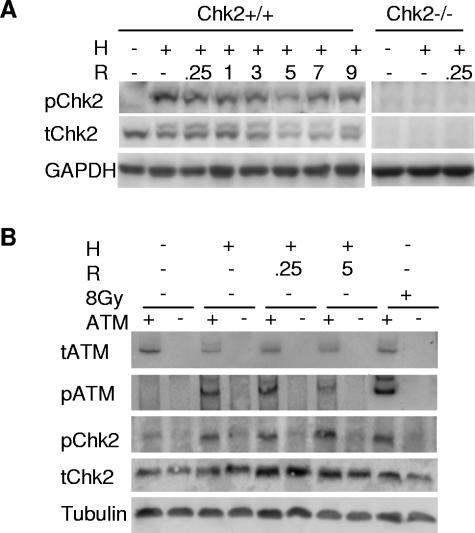
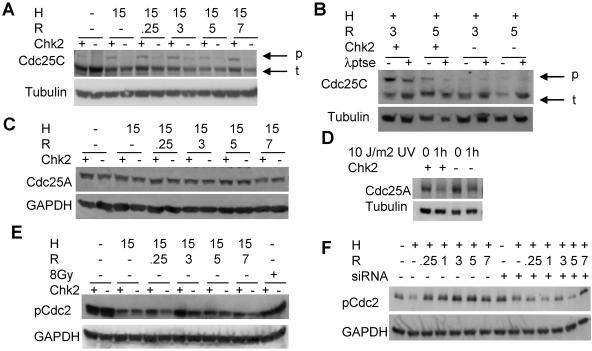
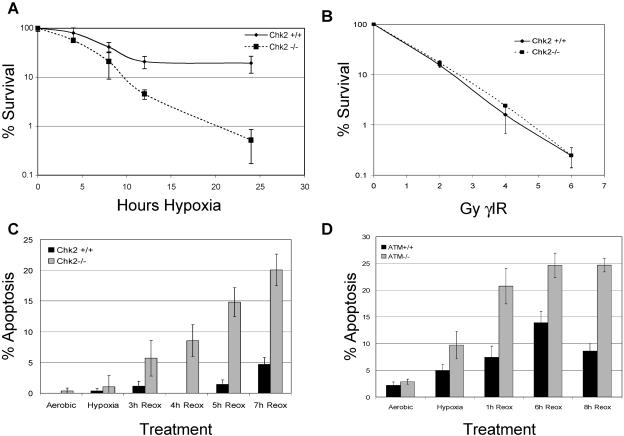
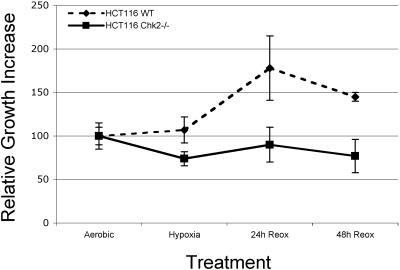
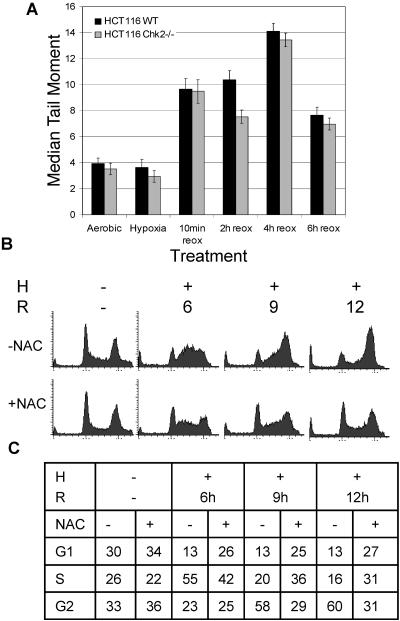
Due to the abnormal vasculature of solid tumors, tumor cell oxygenation can change rapidly with the opening and closing of blood vessels, leading to the activation of both hypoxic response pathways and oxidative stress pathways upon reoxygenation. Here, we report that ataxia telangiectasia mutated-dependent phosphorylation and activation of Chk2 occur in the absence of DNA damage during hypoxia and are maintained during reoxygenation in response to DNA damage. Our studies involving oxidative damage show that Chk2 is required for G2 arrest. Following exposure to both hypoxia and reoxygenation, Chk2-/- cells exhibit an attenuated G2 arrest, increased apoptosis, reduced clonogenic survival, and deficient phosphorylation of downstream targets. These studies indicate that the combination of hypoxia and reoxygenation results in a G2 checkpoint response that is dependent on the tumor suppressor Chk2 and that this checkpoint response is essential for tumor cell adaptation to changes that result from the cycling nature of hypoxia and reoxygenation found in solid tumors.
Figures
References
-
- Abraham, R. T. 2001. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15:2177-2196. - PubMed
-
- Ahn, J. Y., J. K. Schwarz, H. Piwnica-Worms, and C. E. Canman. 2000. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 60:5934-5936. - PubMed
-
- Bakkenist, C. J., and M. B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499-506. - PubMed
-
- Bartek, J., J. Falck, and J. Lukas. 2001. CHK2 kinase-a busy messenger. Nat Rev. Mol. Cell Biol. 2:877-886. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials