

The molecular architecture of the metalloprotease FtsH
- PMID: 16484367
- PMCID: PMC1413944
- DOI: 10.1073/pnas.0600031103
The molecular architecture of the metalloprotease FtsH
Abstract
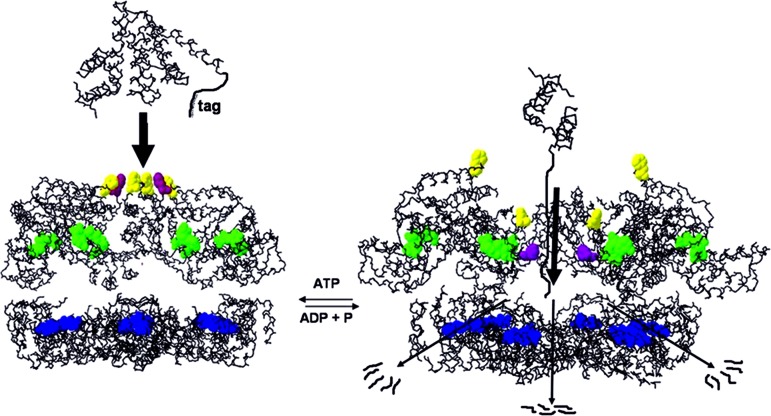
The ATP-dependent integral membrane protease FtsH is universally conserved in bacteria. Orthologs exist in chloroplasts and mitochondria, where in humans the loss of a close FtsH-homolog causes a form of spastic paraplegia. FtsH plays a crucial role in quality control by degrading unneeded or damaged membrane proteins, but it also targets soluble signaling factors like sigma(32) and lambda-CII. We report here the crystal structure of a soluble FtsH construct that is functional in caseinolytic and ATPase assays. The molecular architecture of this hexameric molecule consists of two rings where the protease domains possess an all-helical fold and form a flat hexagon that is covered by a toroid built by the AAA domains. The active site of the protease classifies FtsH as an Asp-zincin, contrary to a previous report. The different symmetries of protease and AAA rings suggest a possible translocation mechanism of the target polypeptide chain into the interior of the molecule where the proteolytic sites are located.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
