

Forced vital capacity (FVC) as an indicator of survival and disease progression in an ALS clinic population
- PMID: 16484652
- PMCID: PMC2077717
- DOI: 10.1136/jnnp.2005.072660
Forced vital capacity (FVC) as an indicator of survival and disease progression in an ALS clinic population
Abstract
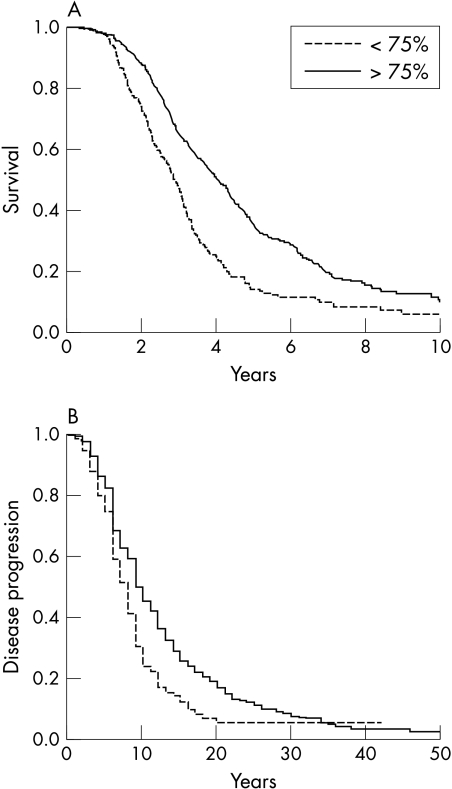
In a large cohort of 1034 patients with the diagnosis of definite or probable amyotrophic lateral sclerosis (ALS), the association of forced vital capacity (FVC) at baseline with (a) time to progression of 20 points in Appel ALS (AALS) score or (b) tracheostomy free survival was investigated. The median survival of ALS patients with baseline FVC <75% was 2.91 years, compared with 4.08 years for patients with baseline FVC >75% (p<0.001). Patients with baseline FVC <75% progressed more rapidly (taking 8.0 months to progress 20 AALS points) compared with patients with baseline FVC >75% (10.0 months, p<0.001). Moreover, FVC at first examination was identified as a significant predictor of survival and disease progression in both univariate and multivariate Cox regression models, after adjustment for age, sex, site of onset, diagnostic delay, riluzole therapy, and use of bilateral positive airway pressure and percutaneous endoscopic gastrostomy (p<0.001). We conclude that a single FVC value obtained at an initial visit may serve as a clinically meaningful predictor of survival and disease progression in ALS.
Conflict of interest statement
Competing interests: none
References
-
- Traynor B J, Zhang H, Shefner J M.et al Functional outcome measures as clinical trial endpoints in ALS. Neurology 2004631933–1935. - PubMed
-
- Caroscio J T, Mulvihill M N, Sterling R.et al Amyotrophic lateral sclerosis: its natural history. Neurol Clin 198751–8. - PubMed
-
- Kleopa K A, Sherman M, Neal B.et al Bipap improves survival and rate of pulmonary function decline in patients with ALS. J Neurol Sci 199916482–88. - PubMed
-
- Haverkamp L J, Appel V, Appel S H. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain 1995118707–719. - PubMed
-
- Miller R G, Munsat T L, Swash M.et al Consensus guidelines for the design and implementation of clinical trials in ALS. J Neurol Sci 19991692–12. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous