

A pooling-deconvolution strategy for biological network elucidation
- PMID: 16489335
- PMCID: PMC2803036
- DOI: 10.1038/nmeth859
A pooling-deconvolution strategy for biological network elucidation
Abstract
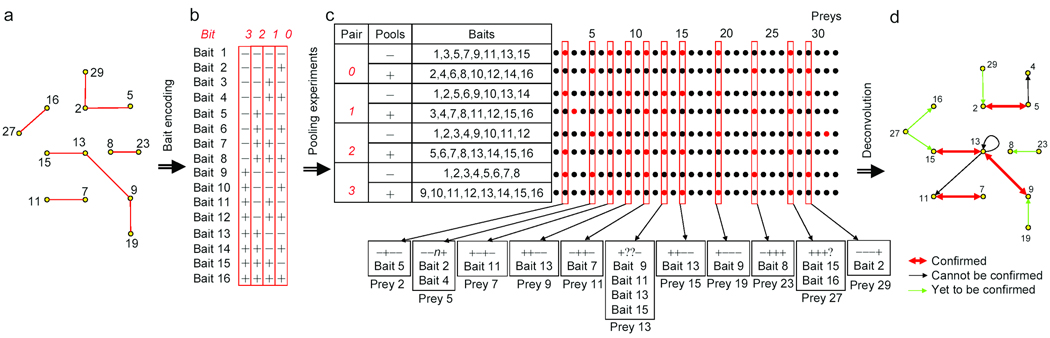
The generation of large-scale data sets is a fundamental requirement of systems biology. But despite recent advances, generation of such high-coverage data remains a major challenge. We developed a pooling-deconvolution strategy that can dramatically decrease the effort required. This strategy, pooling with imaginary tags followed by deconvolution (PI-deconvolution), allows the screening of 2(n) probe proteins (baits) in 2 x n pools, with n replicates for each bait. Deconvolution of baits with their binding partners (preys) can be achieved by reading the prey's profile from the 2 x n experiments. We validated this strategy for protein-protein interaction mapping using both proteome microarrays and a yeast two-hybrid array, demonstrating that PI-deconvolution can be used to identify interactions accurately with fewer experiments and better coverage. We also show that PI-deconvolution can be used to identify protein-small molecule interactions inferred from profiling the yeast deletion collection. PI-deconvolution should be applicable to a wide range of library-against-library approaches and can also be used to optimize array designs.
Figures



Comment in
-
Pooling in systems biology becomes smart.Nat Methods. 2006 Mar;3(3):161-2. doi: 10.1038/nmeth0306-161. Nat Methods. 2006. PMID: 16489330 No abstract available.
References
-
- Phizicky E, Bastiaens PI, Zhu H, Snyder M, Fields S. Protein analysis on a proteomic scale. Nature. 2003;422:208–215. - PubMed
-
- Uetz P, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. - PubMed
-
- Giot L, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
