

TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE
- PMID: 16492804
- PMCID: PMC2118244
- DOI: 10.1084/jem.20052438
TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE
Abstract
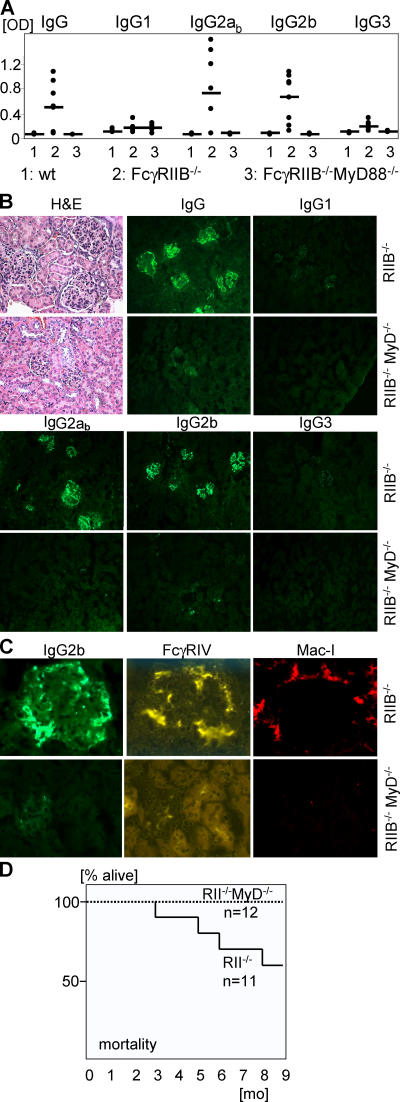
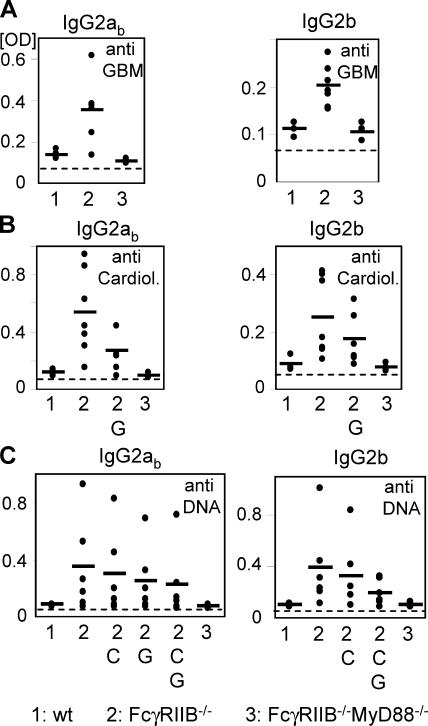
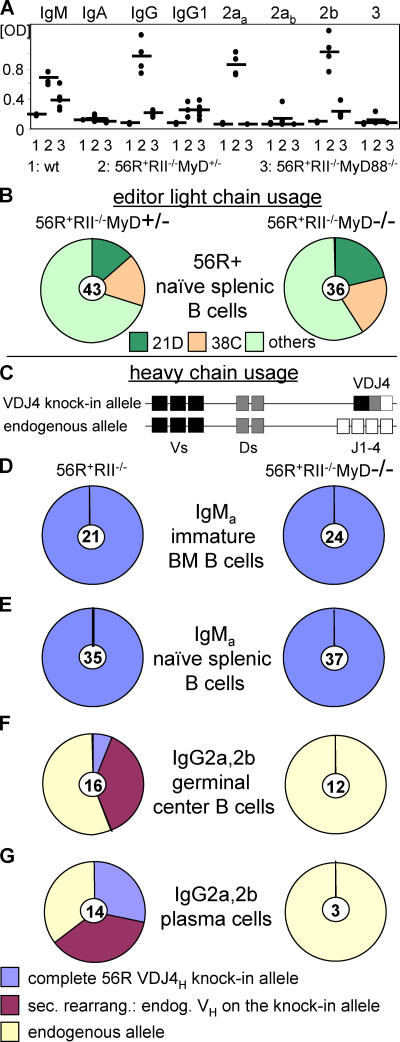
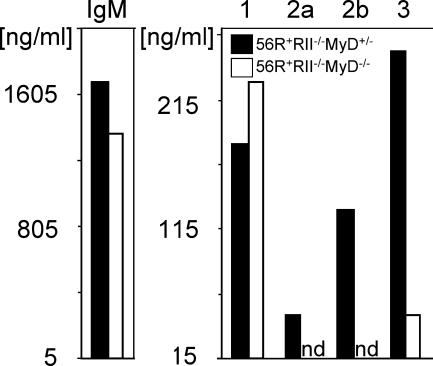
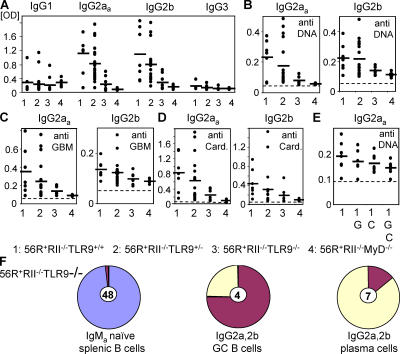
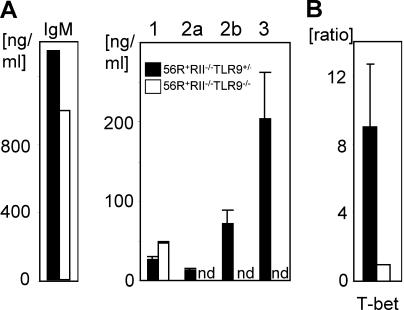
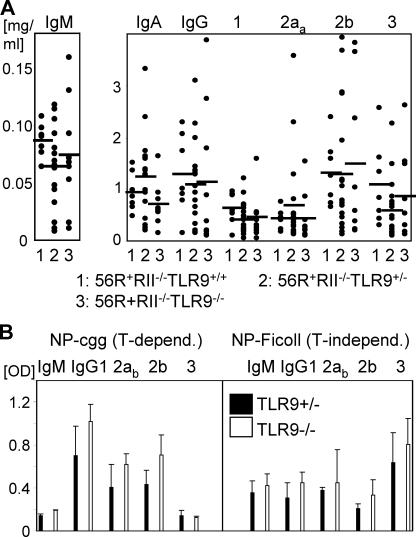
Loss of tolerance in systemic lupus erythematosus (SLE) leads to the generation of autoantibodies, which accumulate in end-organs where they induce disease. Here we show that immunoglobulin (Ig)G2a and 2b autoantibodies are the pathogenic isotypes by recruiting FcgammaRIV expressing macrophages. Class switching, but not development, of IgM anti-self B cells to these pathogenic subclasses requires the innate immune receptor Toll-like receptor (TLR)9 and MyD88 signaling. In their absence, switching of autoreactive B cells to the IgG2a and 2b subclasses is blocked, resulting in reduced pathology and mortality. In contrast, switching of anti-self B cells to IgG1 is not perturbed and generation of nonautoreactive IgG2a and 2b antibodies is not impaired in TLR9-deficient mice. Thus, the TLR9 pathway is a potential target for therapeutic intervention in SLE.
Figures
References
-
- Peter, J.B., and Y. Shoenfeld. 1996. Autoantibodies. Elsevier, New York. 910 pp.
-
- Bolland, S., and J.V. Ravetch. 2000. Spontaneous autoimmune disease in FcγRII deficient mice results from strain-specific epistasis. Immunity. 13:277–285. - PubMed
-
- Rifkin, I.R., and A. Marshak-Rothstein. 2003. T-bet: the Toll-bridge to class-switch recombination? Nat. Immunol. 4:650–652. - PubMed
-
- Fields, M.L., and J. Erikson. 2003. The regulation of lupus-associated autoantibodies: immunoglobulin transgenic models. Curr. Opin. Immunol. 15:709–717. - PubMed
-
- Wakeland, E.K., K. Liu, R.R. Graham, and T.W. Behrens. 2001. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 15:397–408. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
