

RNA secondary structure prediction from sequence alignments using a network of k-nearest neighbor classifiers
- PMID: 16495232
- PMCID: PMC1383574
- DOI: 10.1261/rna.2164906
RNA secondary structure prediction from sequence alignments using a network of k-nearest neighbor classifiers
Abstract
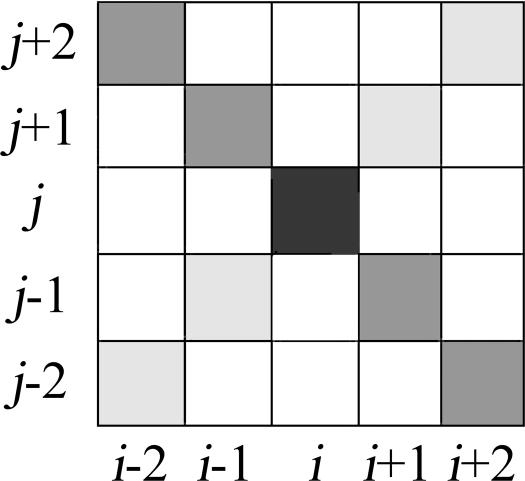
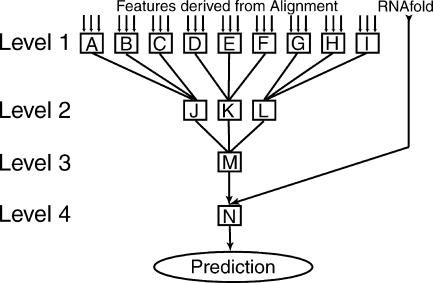
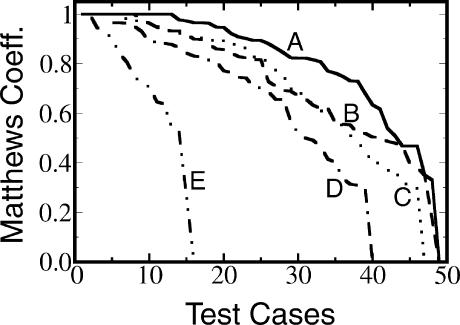
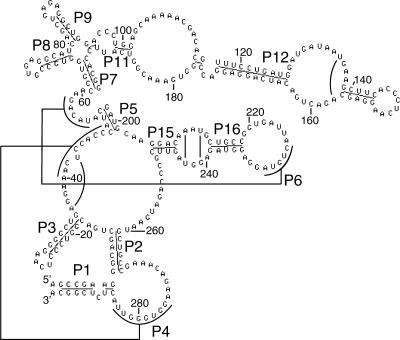
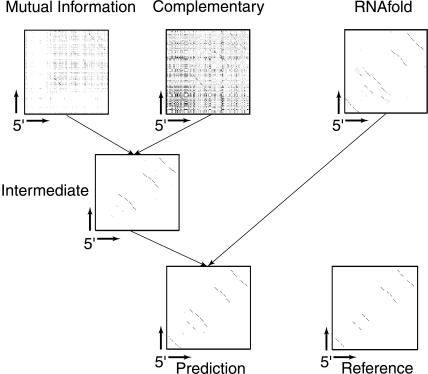
We present a machine learning method (a hierarchical network of k-nearest neighbor classifiers) that uses an RNA sequence alignment in order to predict a consensus RNA secondary structure. The input to the network is the mutual information, the fraction of complementary nucleotides, and a novel consensus RNAfold secondary structure prediction of a pair of alignment columns and its nearest neighbors. Given this input, the network computes a prediction as to whether a particular pair of alignment columns corresponds to a base pair. By using a comprehensive test set of 49 RFAM alignments, the program KNetFold achieves an average Matthews correlation coefficient of 0.81. This is a significant improvement compared with the secondary structure prediction methods PFOLD and RNAalifold. By using the example of archaeal RNase P, we show that the program can also predict pseudoknot interactions.
Figures
References
-
- Akmaev, V.R., Kelley, S.T., and Stormo, G.D. 2000. Phylogeneticically enhanced statistical tools for RNA structure prediction. Bioinformatics 16: 501–512. - PubMed
-
- Arya, S. and Mount, D.M. 1993. Algorithms for fast vector quantization. Proceedings of DCC ’93: Data compression conference (eds. J.A. Storer, and M. Cohn), pp. 381–390. IEEE Press, Snowbird, UT.
-
- Baldi, P., Brunak, S., Chauvin, Y., Anderson, C., and Nielsen, H. 2000. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 16: 412–424. - PubMed
-
- Basharin, G.P. 1959. On a statistical estimate for the entropy of a sequence of independent random variables. Theory Probability Appl. 4: 333–336.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases