

Automated extraction of backbone deuteration levels from amide H/2H mass spectrometry experiments
- PMID: 16501228
- PMCID: PMC2249778
- DOI: 10.1110/ps.051774906
Automated extraction of backbone deuteration levels from amide H/2H mass spectrometry experiments
Abstract
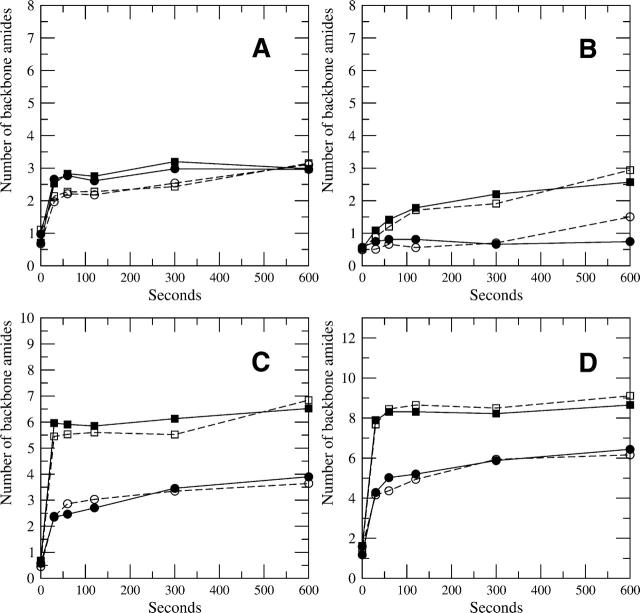
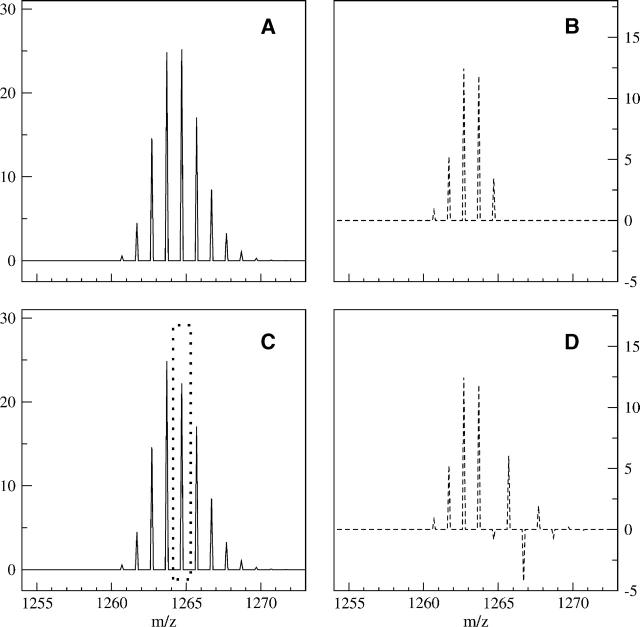
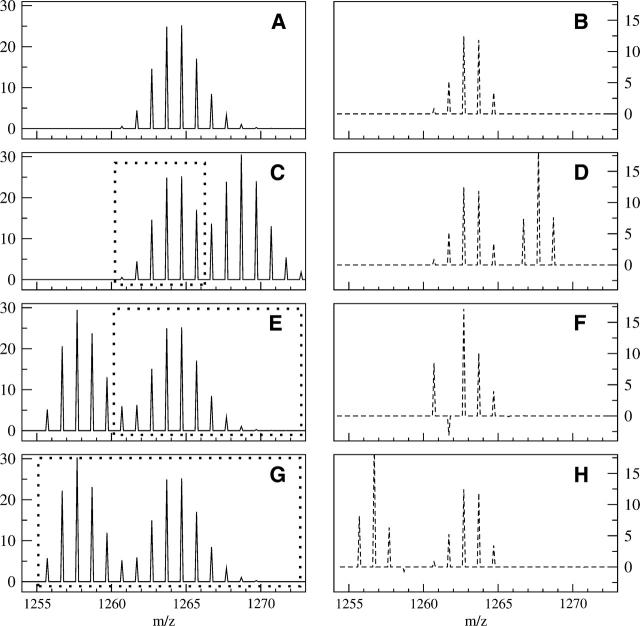
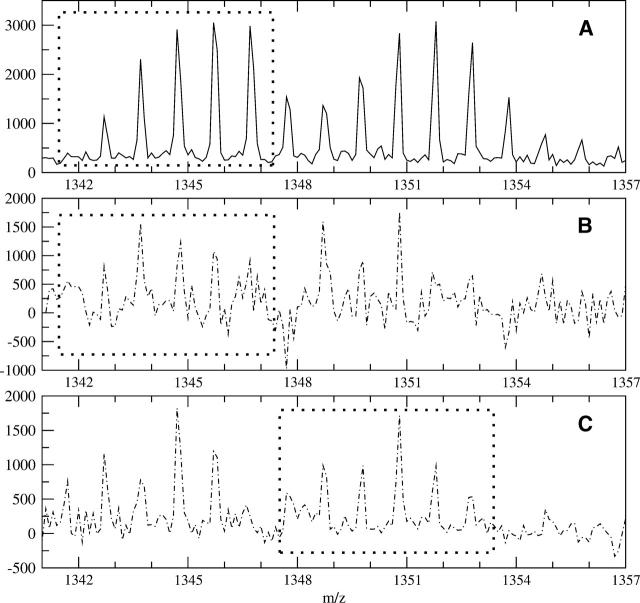
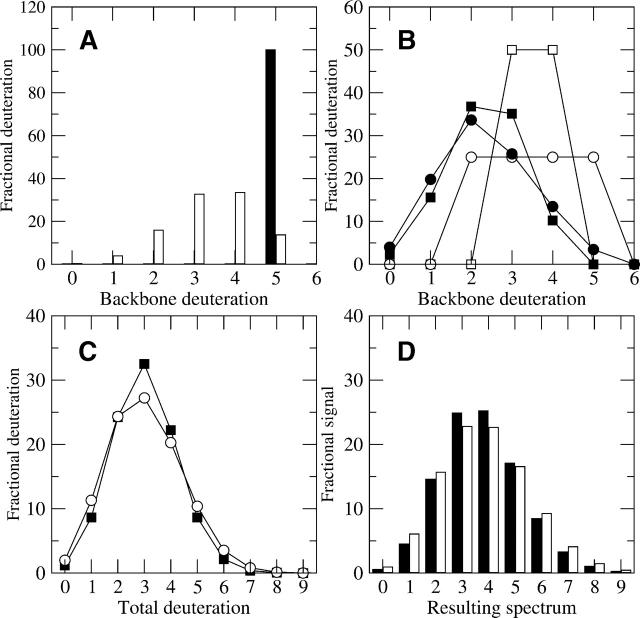
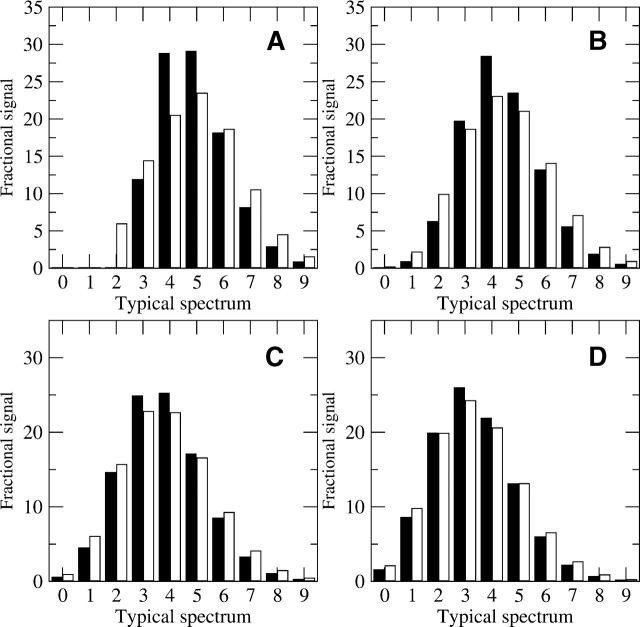
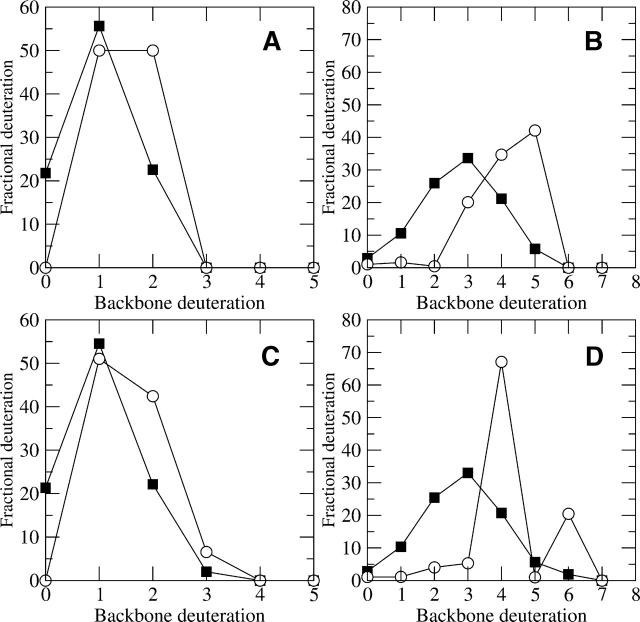
A Fourier deconvolution method has been developed to explicitly determine the amount of backbone amide deuterium incorporated into protein regions or segments by hydrogen/deuterium (H/D) exchange with high-resolution mass spectrometry. Determination and analysis of the level and number of backbone amide exchanging in solution provide more information about the solvent accessibility of the protein than do previous centroid methods, which only calculate the average deuterons exchanged. After exchange, a protein is digested into peptides as a way of determining the exchange within a local area of the protein. The mass of a peptide upon deuteration is a sum of the natural isotope abundance, fast exchanging side-chain hydrogens (present in MALDI-TOF H/2H data) and backbone amide exchange. Removal of the components of the isotopic distribution due to the natural isotope abundances and the fast exchanging side-chains allows for a precise quantification of the levels of backbone amide exchange, as is shown by an example from protein kinase A. The deconvoluted results are affected by overlapping peptides or inconsistent mass envelopes, and evaluation procedures for these cases are discussed. Finally, a method for determining the back exchange corrected populations is presented, and its effect on the data is discussed under various circumstances.
Figures
References
-
- Anand, G.S., Hughes, C.A., Jones, J.M., Taylor, S.S., and Komives, E.A. 2002. Amide H/2H exchange reveals communication between the cAMP and catalytic subunit-binding sites in the RIαsubunit of protein kinase A. J. Mol. Biol. 323: 377–386. - PubMed
-
- Clauser, K.R., Baker, P., and Burlingame, A.L. 1999. Role of accurate mass measurement (± 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal. Chem. 71: 2871–2882. - PubMed
-
- Dempsey, C.E. 2001. Hydrogen exchange in peptides and proteins using NMR spectroscopy. Progr. Nuclear Magn. Reson. Spectr. 39: 135–170.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
