

Biclustering of gene expression data by Non-smooth Non-negative Matrix Factorization
- PMID: 16503973
- PMCID: PMC1434777
- DOI: 10.1186/1471-2105-7-78
Biclustering of gene expression data by Non-smooth Non-negative Matrix Factorization
Abstract
Background: The extended use of microarray technologies has enabled the generation and accumulation of gene expression datasets that contain expression levels of thousands of genes across tens or hundreds of different experimental conditions. One of the major challenges in the analysis of such datasets is to discover local structures composed by sets of genes that show coherent expression patterns across subsets of experimental conditions. These patterns may provide clues about the main biological processes associated to different physiological states.
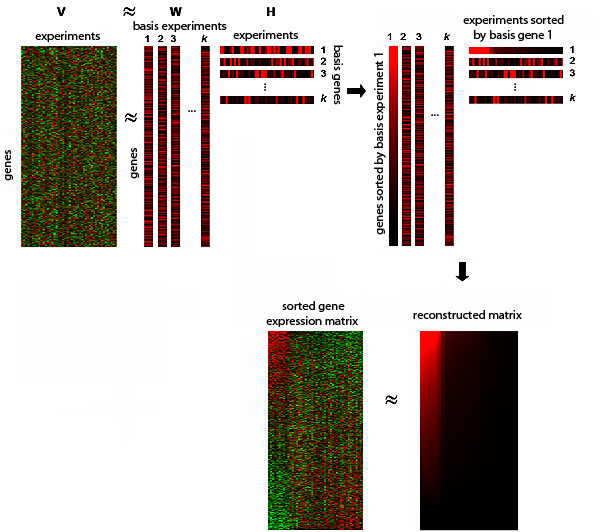
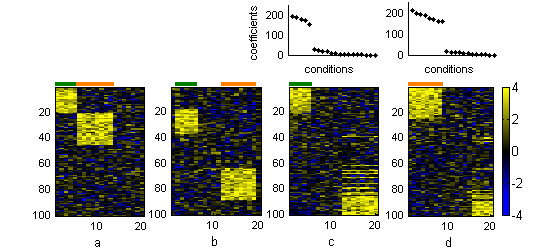
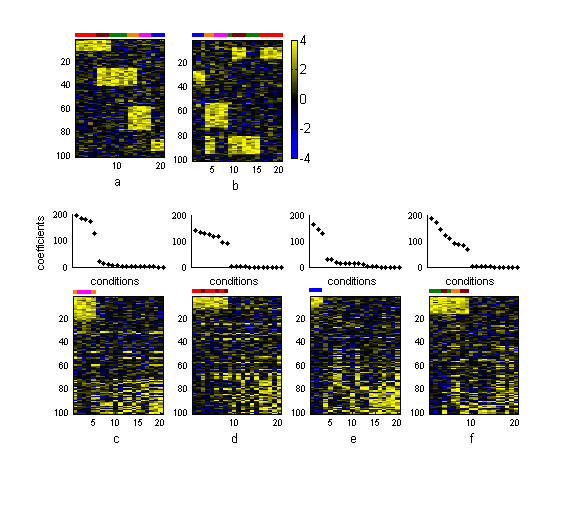
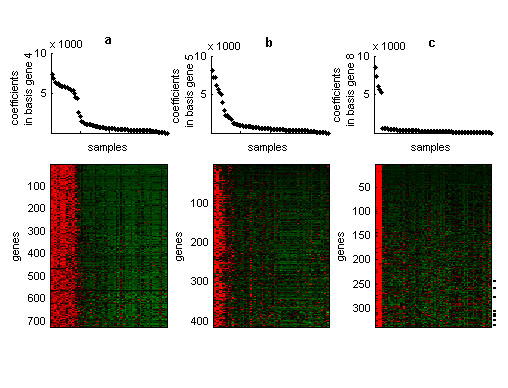
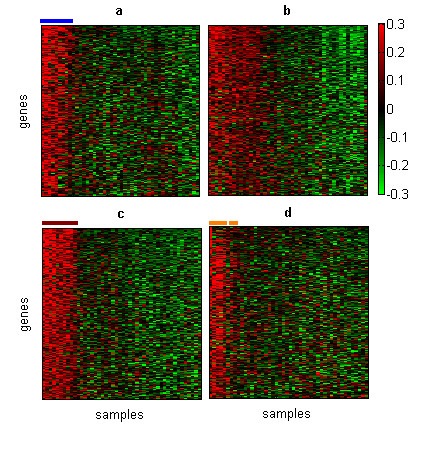
Results: In this work we present a methodology able to cluster genes and conditions highly related in sub-portions of the data. Our approach is based on a new data mining technique, Non-smooth Non-Negative Matrix Factorization (nsNMF), able to identify localized patterns in large datasets. We assessed the potential of this methodology analyzing several synthetic datasets as well as two large and heterogeneous sets of gene expression profiles. In all cases the method was able to identify localized features related to sets of genes that show consistent expression patterns across subsets of experimental conditions. The uncovered structures showed a clear biological meaning in terms of relationships among functional annotations of genes and the phenotypes or physiological states of the associated conditions.
Conclusion: The proposed approach can be a useful tool to analyze large and heterogeneous gene expression datasets. The method is able to identify complex relationships among genes and conditions that are difficult to identify by standard clustering algorithms.
Figures
References
-
- Stoughton RB. Applications of DNA Microarrays in Biology. Annu Rev Biochem. 2004 - PubMed
-
- Hsiao LL, Dangond F, Yoshida T, Hong R, Jensen RV, Misra J, Dillon W, Lee KF, Clark KE, Haverty P, Weng Z, Mutter GL, Frosch MP, Macdonald ME, Milford EL, Crum CP, Bueno R, Pratt RE, Mahadevappa M, Warrington JA, Stephanopoulos G, Gullans SR. A compendium of gene expression in normal human tissues. Physiol Genomics. 2001;7:97–104. - PubMed
-
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, Kidd MJ, King AM, Meyer MR, Slade D, Lum PY, Stepaniants SB, Shoemaker DD, Gachotte D, Chakraburtty K, Simon J, Bard M, Friend SH. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/S0092-8674(00)00015-5. - DOI - PubMed
-
- Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, Patapoutian A, Hampton GM, Schultz PG, Hogenesch JB. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
