

Endotoxin induced peritonitis elicits monocyte immigration into the lung: implications on alveolar space inflammatory responsiveness
- PMID: 16503998
- PMCID: PMC1388208
- DOI: 10.1186/1465-9921-7-30
Endotoxin induced peritonitis elicits monocyte immigration into the lung: implications on alveolar space inflammatory responsiveness
Abstract
Background: Acute peritonitis developing in response to gram-negative bacterial infection is known to act as a trigger for the development of acute lung injury which is often complicated by the development of nosocomial pneumonia. We hypothesized that endotoxin-induced peritonitis provokes recruitment of monocytes into the lungs, which amplifies lung inflammatory responses to a second hit intra-alveolar challenge with endotoxin.
Methods: Serum and lavage cytokines as well as bronchoalveolar lavage fluid cells were analyzed at different time points after intraperitoneal or intratracheal application of LPS.
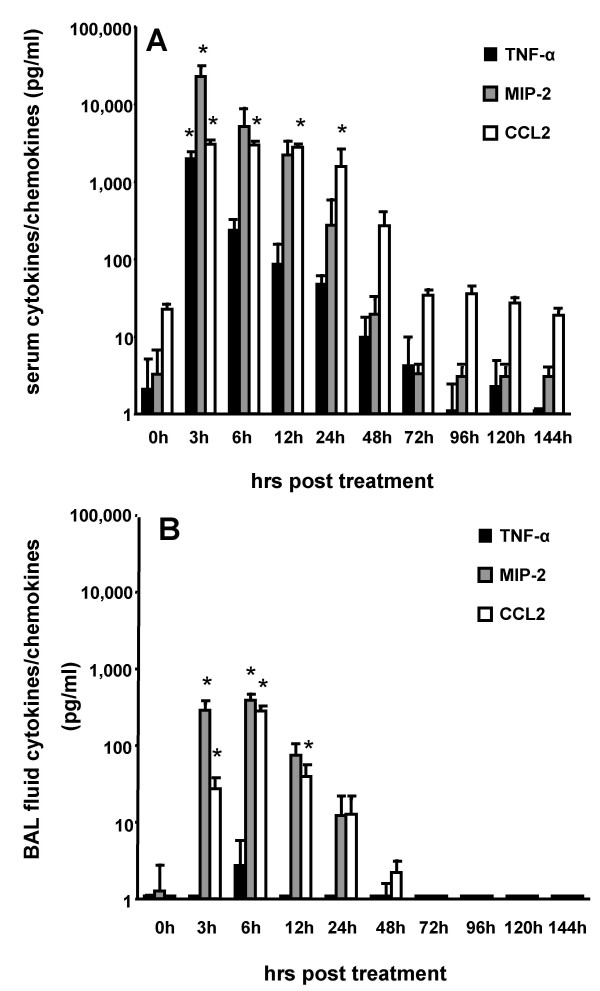
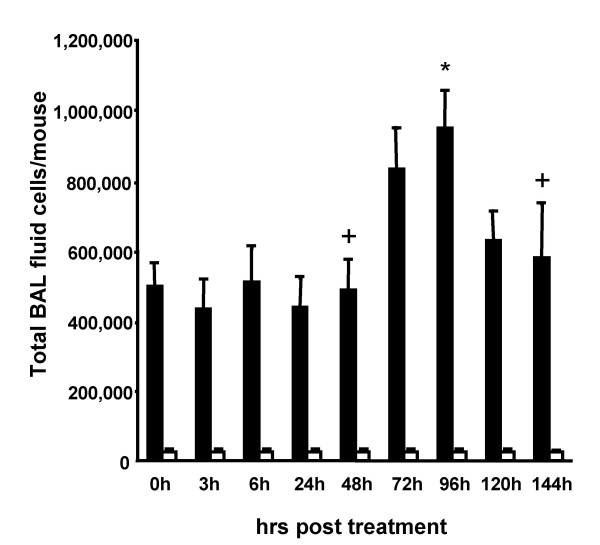
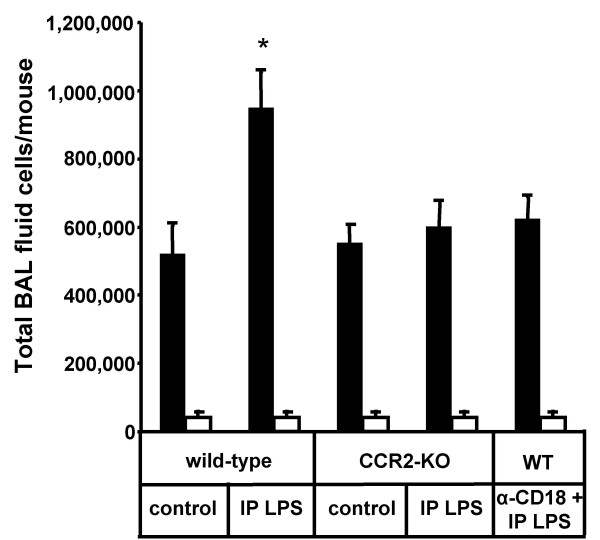
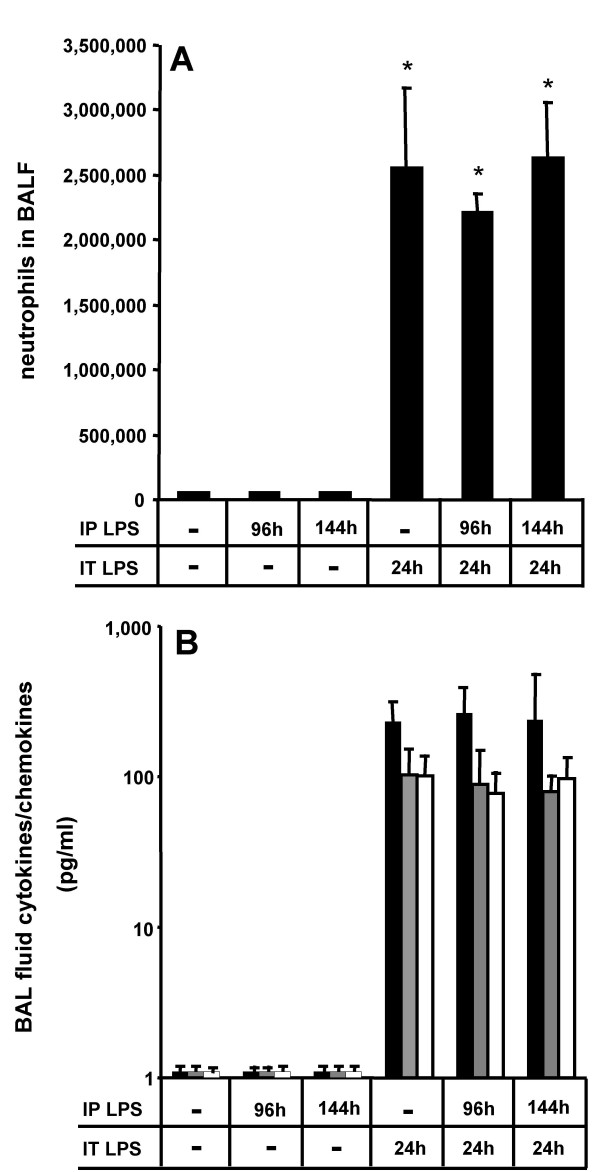
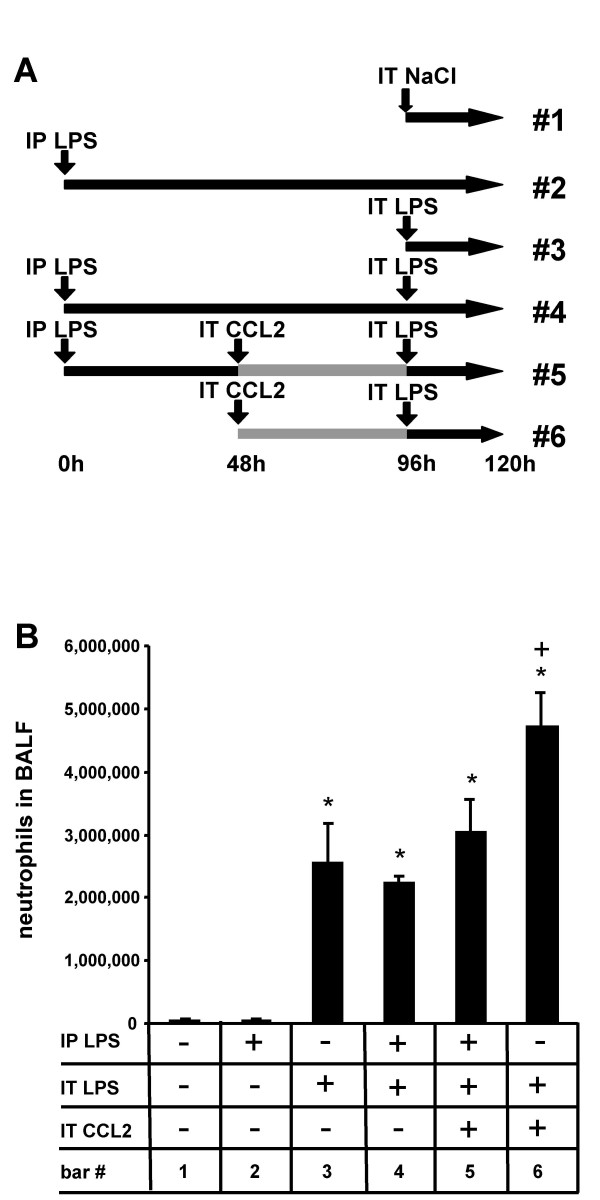
Results: We observed that mice challenged with intraperitoneal endotoxin developed rapidly increasing serum and bronchoalveolar lavage fluid (BALF) cytokine and chemokine levels (TNFalpha, MIP-2, CCL2) and a nearly two-fold expansion of the alveolar macrophage population by 96 h, but this was not associated with the development of neutrophilic alveolitis. In contrast, expansion of the alveolar macrophage pool was not observed in CCR2-deficient mice and in wild-type mice systemically pretreated with the anti-CD18 antibody GAME-46. An intentional two-fold expansion of alveolar macrophage numbers by intratracheal CCL2 following intraperitoneal endotoxin did not exacerbate the development of acute lung inflammation in response to intratracheal endotoxin compared to mice challenged only with intratracheal endotoxin.
Conclusion: These data, taken together, show that intraperitoneal endotoxin triggers a CCR2-dependent de novo recruitment of monocytes into the lungs of mice but this does not result in an accentuation of neutrophilic lung inflammation. This finding represents a previously unrecognized novel inflammatory component of lung inflammation that results from endotoxin-induced peritonitis.
Figures
Similar articles
-
Alveolar JE/MCP-1 and endotoxin synergize to provoke lung cytokine upregulation, sequential neutrophil and monocyte influx, and vascular leakage in mice.Am J Respir Crit Care Med. 2001 Aug 1;164(3):406-11. doi: 10.1164/ajrccm.164.3.2009055. Am J Respir Crit Care Med. 2001. PMID: 11500341
-
CCR2-positive monocytes recruited to inflamed lungs downregulate local CCL2 chemokine levels.Am J Physiol Lung Cell Mol Physiol. 2005 Feb;288(2):L350-8. doi: 10.1152/ajplung.00061.2004. Epub 2004 Oct 29. Am J Physiol Lung Cell Mol Physiol. 2005. PMID: 15516494
-
The role of CC chemokine receptor 2 in alveolar monocyte and neutrophil immigration in intact mice.Am J Respir Crit Care Med. 2002 Aug 1;166(3):268-73. doi: 10.1164/rccm.2112012. Am J Respir Crit Care Med. 2002. PMID: 12153956
-
Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis.J Immunol. 2003 Mar 15;170(6):3273-8. doi: 10.4049/jimmunol.170.6.3273. J Immunol. 2003. PMID: 12626586
-
Alveolar macrophages regulate neutrophil recruitment in endotoxin-induced lung injury.Respir Res. 2005 Jun 22;6(1):61. doi: 10.1186/1465-9921-6-61. Respir Res. 2005. PMID: 15972102 Free PMC article.
Cited by
-
Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones.PLoS One. 2012;7(2):e32366. doi: 10.1371/journal.pone.0032366. Epub 2012 Feb 28. PLoS One. 2012. PMID: 22389696 Free PMC article.
-
Hepatocyte SHP-1 is a Critical Modulator of Inflammation During Endotoxemia.Sci Rep. 2017 May 22;7(1):2218. doi: 10.1038/s41598-017-02512-7. Sci Rep. 2017. PMID: 28533521 Free PMC article.
-
Lipopolysaccharide modulates neutrophil recruitment and macrophage polarization on lymphatic vessels and impairs lymphatic function in rat mesentery.Am J Physiol Heart Circ Physiol. 2015 Dec 15;309(12):H2042-57. doi: 10.1152/ajpheart.00467.2015. Epub 2015 Oct 9. Am J Physiol Heart Circ Physiol. 2015. PMID: 26453331 Free PMC article.
-
Decreased Risk of Ventilator-Associated Pneumonia in Sepsis Due to Intra-Abdominal Infection.PLoS One. 2015 Sep 4;10(9):e0137262. doi: 10.1371/journal.pone.0137262. eCollection 2015. PLoS One. 2015. PMID: 26339904 Free PMC article.
-
Low Density Lipoprotein Receptor-Related Protein-1 in Cardiac Inflammation and Infarct Healing.Front Cardiovasc Med. 2019 Apr 26;6:51. doi: 10.3389/fcvm.2019.00051. eCollection 2019. Front Cardiovasc Med. 2019. PMID: 31080804 Free PMC article. Review.
References
-
- Hirano S. Migratory responses of PMN after intraperitoneal and intratracheal administration of lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol. 1996;270:L836–L845. - PubMed
-
- Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, Kunkel SL, Walz A, Hudson LD, Martin TR. Inflammatory cytokines in patients with persistence of the akute respiratory distress syndrome. Am J Respir Crit Care Med. 1996;154:602–611. - PubMed
-
- Rosseau S, Hammerl P, Maus U, Walmrath HD, Schütte H, Grimminger F, Seeger W, Lohmeyer J. Phenotypic characterization of alveolar monocyte recruitment in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2000;279:L25–L35. - PubMed
-
- Maus U, Huwe J, Maus R, Seeger W, Lohmeyer J. Alveolar JE/MCP-1 and endotoxin synergize to provoke lung cytokine upregulation, sequential neutrophil and monocyte influx and vascular leakage in mice. Am J Respir Crit Care Med. 2001;164:406–411. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
