

CARAT: a novel method for allelic detection of DNA copy number changes using high density oligonucleotide arrays
- PMID: 16504045
- PMCID: PMC1402331
- DOI: 10.1186/1471-2105-7-83
CARAT: a novel method for allelic detection of DNA copy number changes using high density oligonucleotide arrays
Abstract
Background: DNA copy number alterations are one of the main characteristics of the cancer cell karyotype and can contribute to the complex phenotype of these cells. These alterations can lead to gains in cellular oncogenes as well as losses in tumor suppressor genes and can span small intervals as well as involve entire chromosomes. The ability to accurately detect these changes is central to understanding how they impact the biology of the cell.
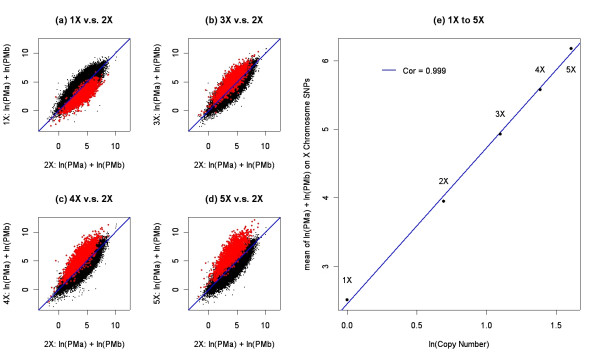
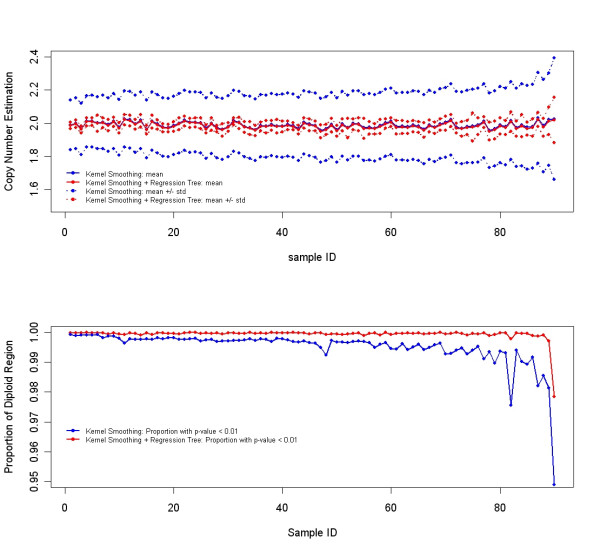
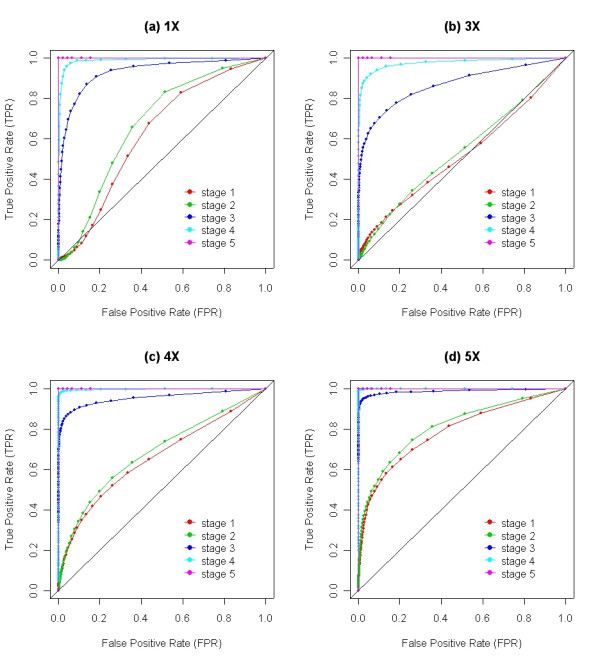
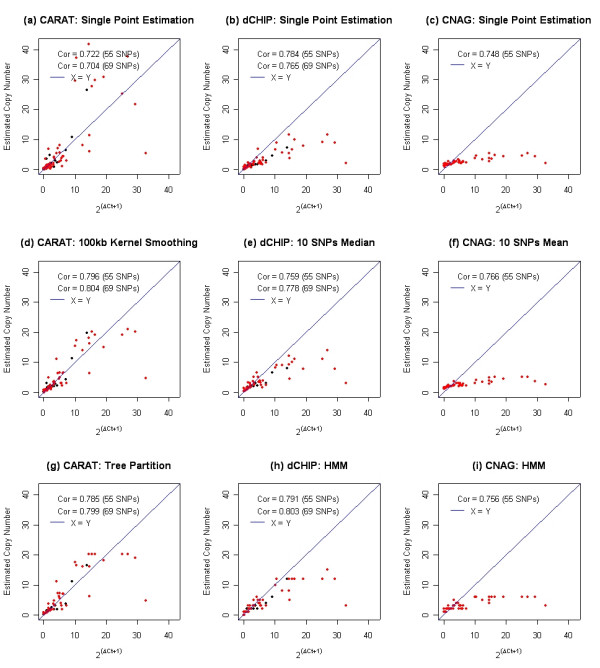
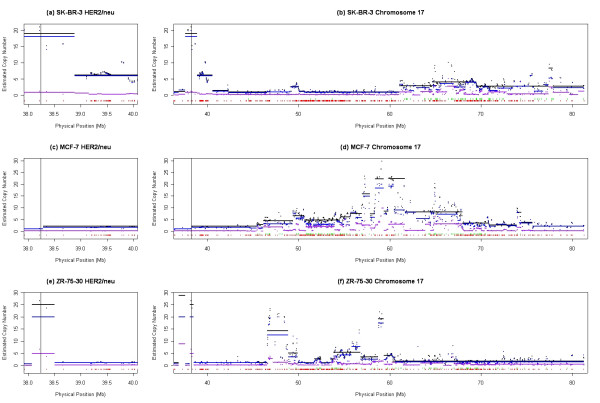
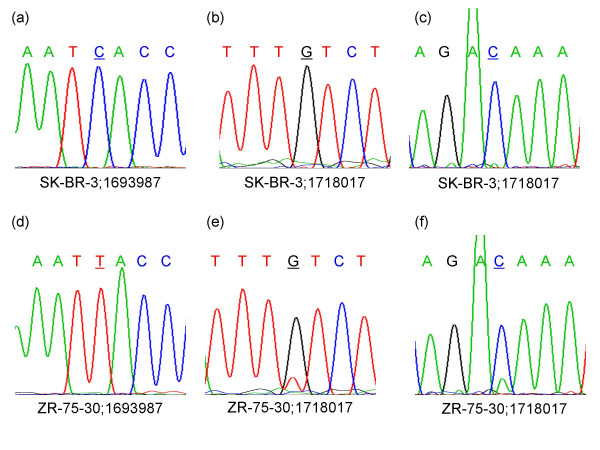
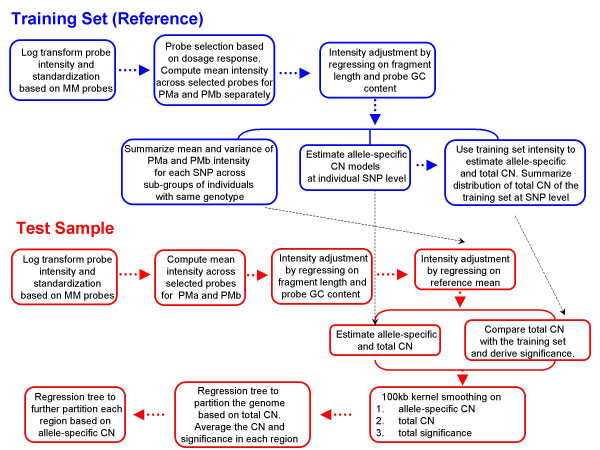
Results: We describe a novel algorithm called CARAT (Copy Number Analysis with Regression And Tree) that uses probe intensity information to infer copy number in an allele-specific manner from high density DNA oligonuceotide arrays designed to genotype over 100,000 SNPs. Total and allele-specific copy number estimations using CARAT are independently evaluated for a subset of SNPs using quantitative PCR and allelic TaqMan reactions with several human breast cancer cell lines. The sensitivity and specificity of the algorithm are characterized using DNA samples containing differing numbers of X chromosomes as well as a test set of normal individuals. Results from the algorithm show a high degree of agreement with results from independent verification methods.
Conclusion: Overall, CARAT automatically detects regions with copy number variations and assigns a significance score to each alteration as well as generating allele-specific output. When coupled with SNP genotype calls from the same array, CARAT provides additional detail into the structure of genome wide alterations that can contribute to allelic imbalance.
Figures
References
-
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
