

Integrating protein structures and precomputed genealogies in the Magnum database: examples with cellular retinoid binding proteins
- PMID: 16504077
- PMCID: PMC1475641
- DOI: 10.1186/1471-2105-7-89
Integrating protein structures and precomputed genealogies in the Magnum database: examples with cellular retinoid binding proteins
Abstract
Background: When accurate models for the divergent evolution of protein sequences are integrated with complementary biological information, such as folded protein structures, analyses of the combined data often lead to new hypotheses about molecular physiology. This represents an excellent example of how bioinformatics can be used to guide experimental research. However, progress in this direction has been slowed by the lack of a publicly available resource suitable for general use.
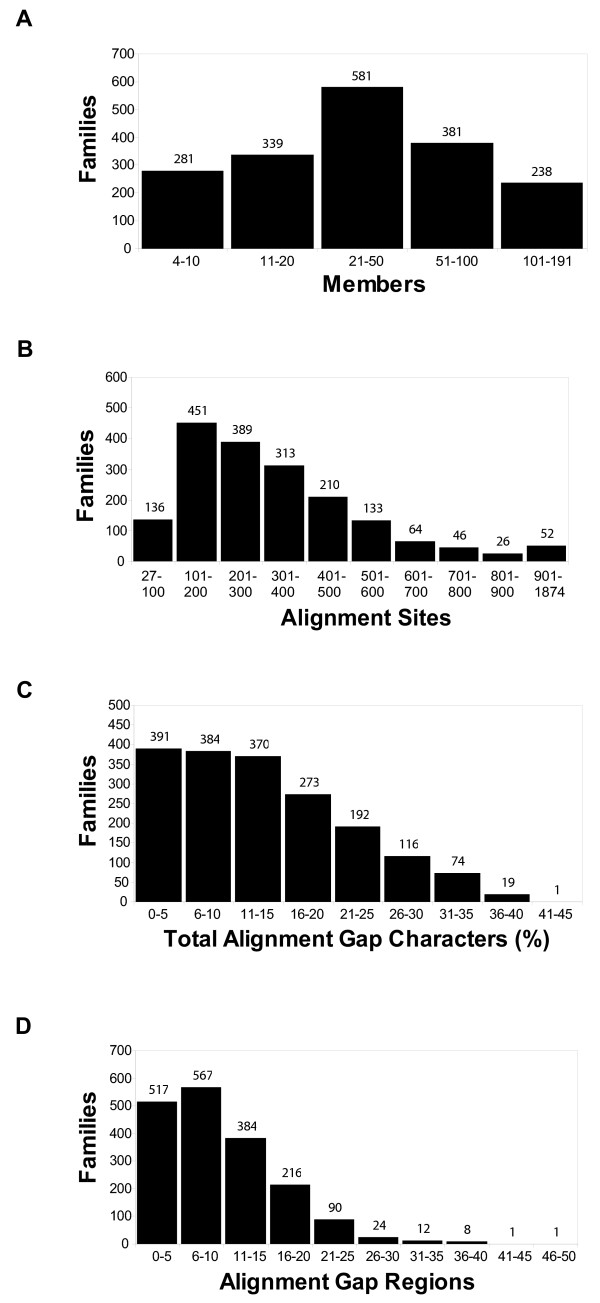
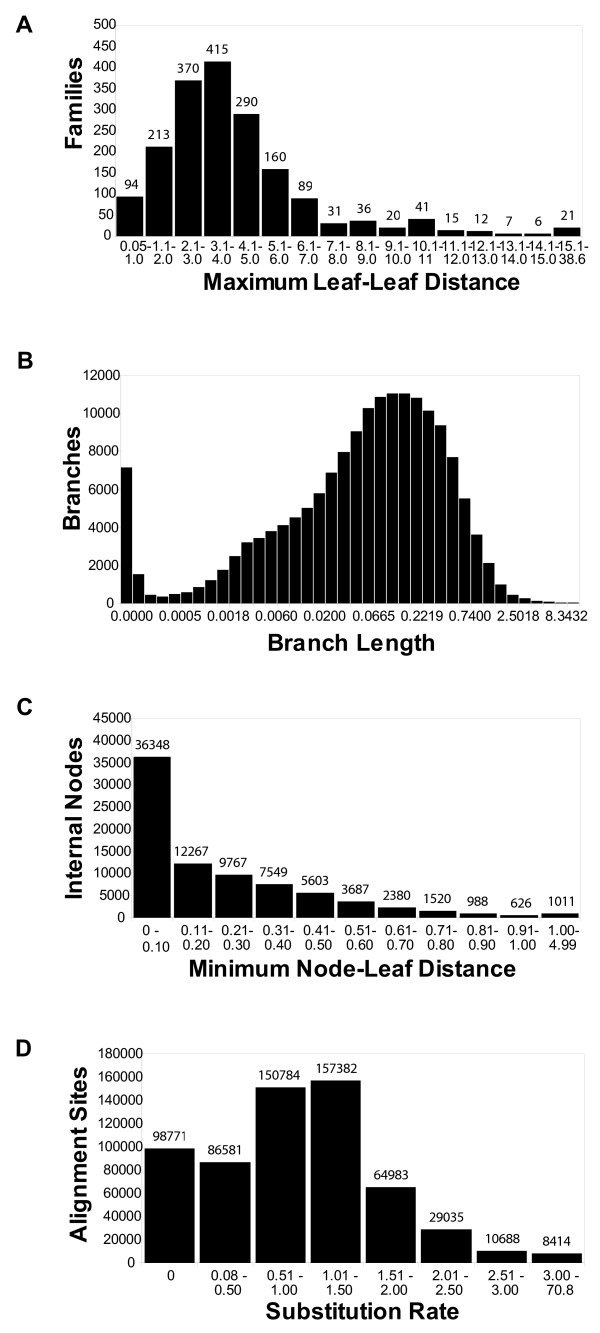
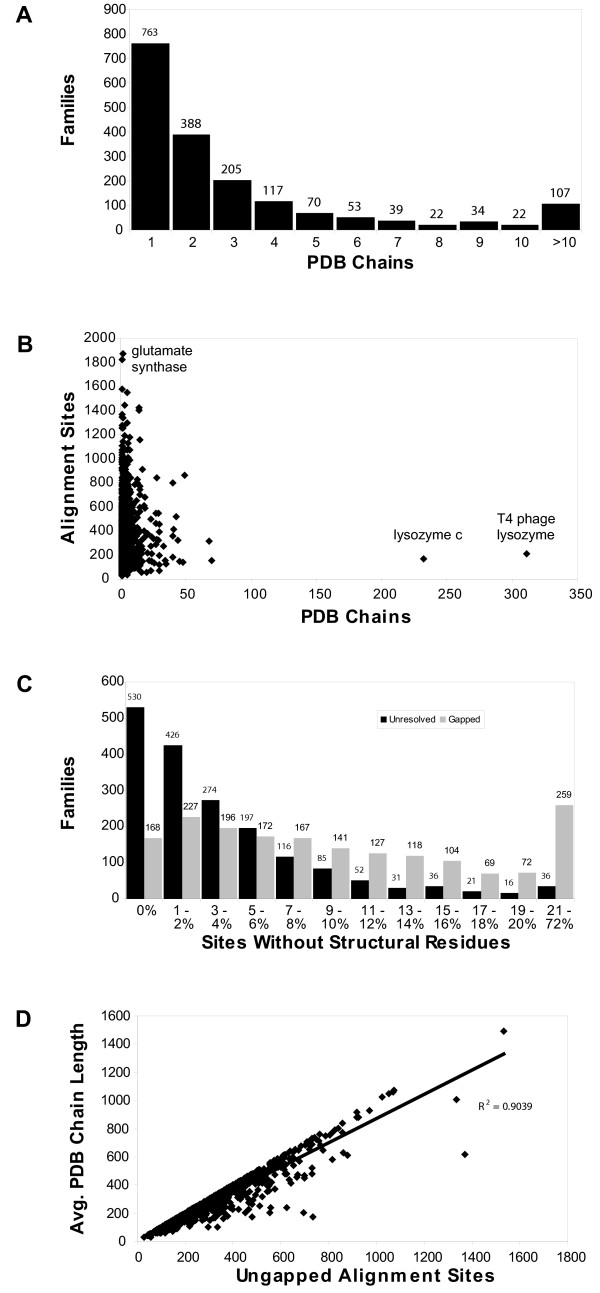
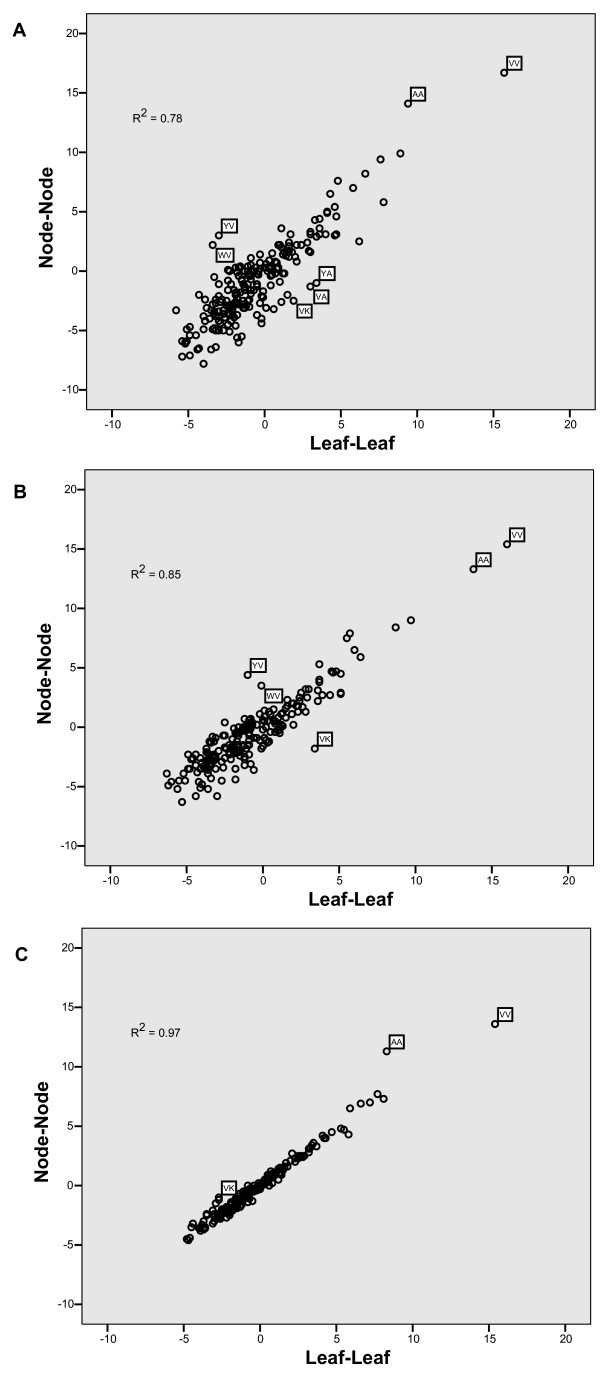
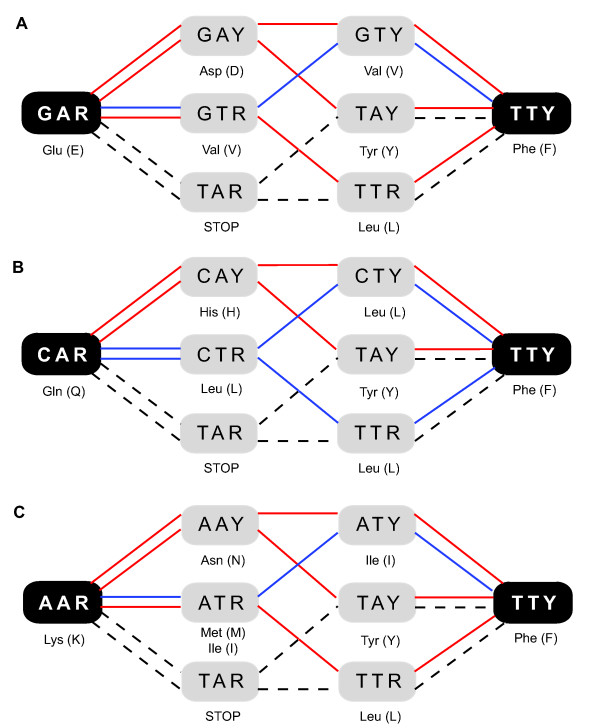
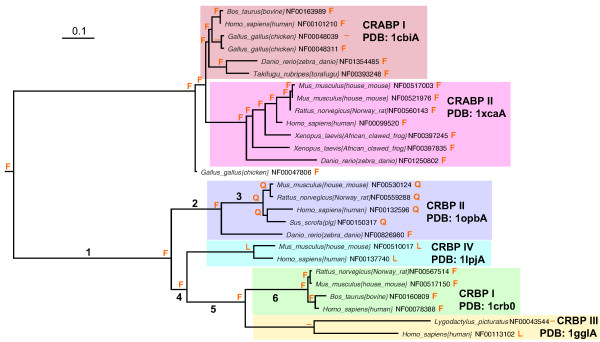
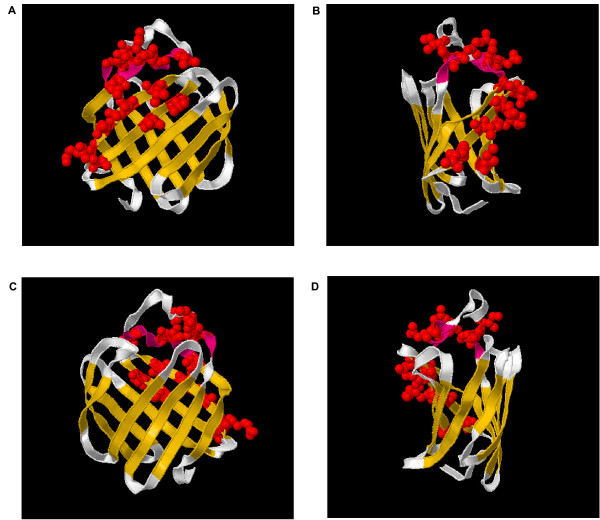
Results: The precomputed Magnum database offers a solution to this problem for ca. 1,800 full-length protein families with at least one crystal structure. The Magnum deliverables include 1) multiple sequence alignments, 2) mapping of alignment sites to crystal structure sites, 3) phylogenetic trees, 4) inferred ancestral sequences at internal tree nodes, and 5) amino acid replacements along tree branches. Comprehensive evaluations revealed that the automated procedures used to construct Magnum produced accurate models of how proteins divergently evolve, or genealogies, and correctly integrated these with the structural data. To demonstrate Magnum's capabilities, we asked for amino acid replacements requiring three nucleotide substitutions, located at internal protein structure sites, and occurring on short phylogenetic tree branches. In the cellular retinoid binding protein family a site that potentially modulates ligand binding affinity was discovered. Recruitment of cellular retinol binding protein to function as a lens crystallin in the diurnal gecko afforded another opportunity to showcase the predictive value of a browsable database containing branch replacement patterns integrated with protein structures.
Conclusion: We integrated two areas of protein science, evolution and structure, on a large scale and created a precomputed database, known as Magnum, which is the first freely available resource of its kind. Magnum provides evolutionary and structural bioinformatics resources that are useful for identifying experimentally testable hypotheses about the molecular basis of protein behaviors and functions, as illustrated with the examples from the cellular retinoid binding proteins.
Figures
References
-
- Taylor WR, Hatrick K. Compensating changes in protein multiple sequence alignments. Protein Eng. 1994;7:341–348. - PubMed
-
- Shindyalov IN, Kolchanov NA, Sander C. Can three-dimensional contacts in protein structures be predicted by analysis of correlated mutations? Protein Eng. 1994;7:349–358. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
