

Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease
- PMID: 16509766
- PMCID: PMC1391977
- DOI: 10.1371/journal.pmed.0030100
Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease
Abstract
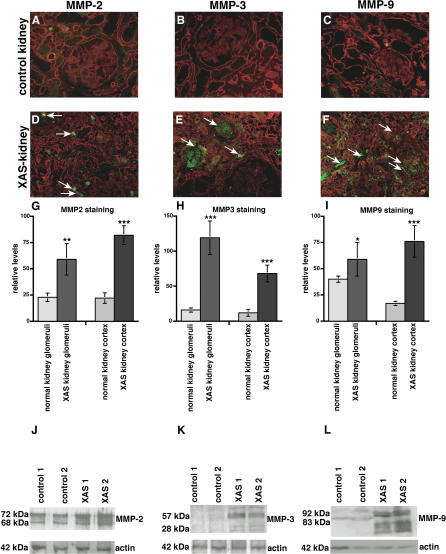
Background: Glomerular basement membrane (GBM), a key component of the blood-filtration apparatus in the in the kidney, is formed through assembly of type IV collagen with laminins, nidogen, and sulfated proteoglycans. Mutations or deletions involving alpha3(IV), alpha4(IV), or alpha5(IV) chains of type IV collagen in the GBM have been identified as the cause for Alport syndrome in humans, a progressive hereditary kidney disease associated with deafness. The pathological mechanisms by which such mutations lead to eventual kidney failure are not completely understood.
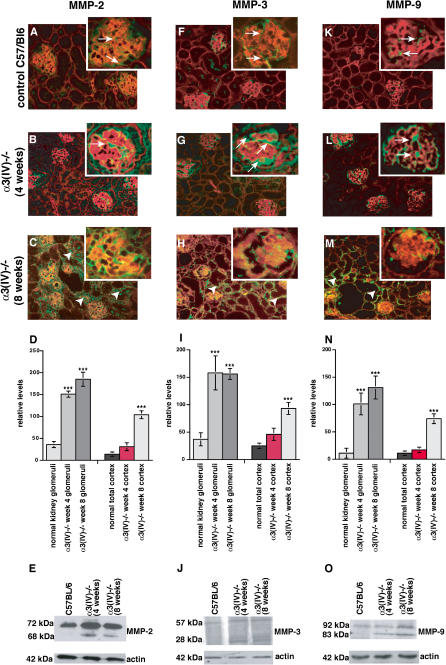
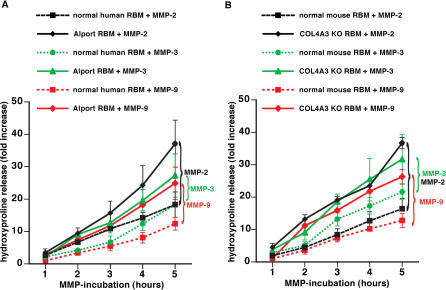
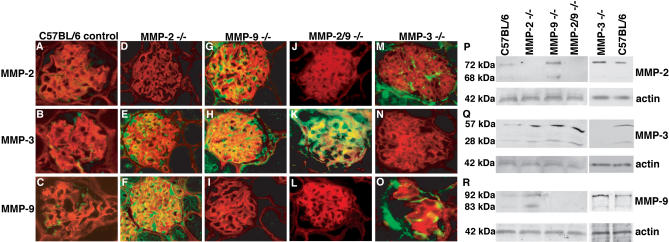
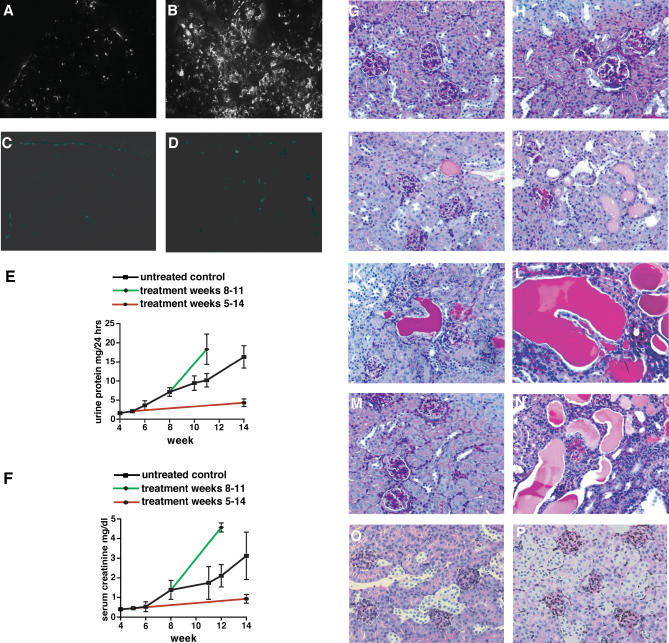
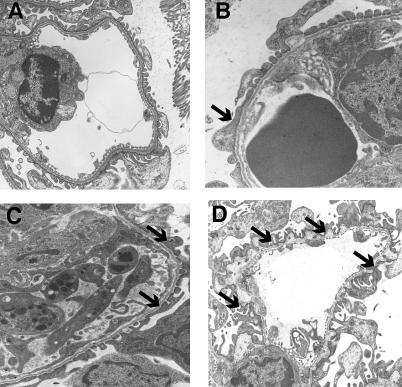
Methods and findings: We showed that increased susceptibility of defective human Alport GBM to proteolytic degradation is mediated by three different matrix metalloproteinases (MMPs)--MMP-2, MMP-3, and MMP-9--which influence the progression of renal dysfunction in alpha3(IV)-/- mice, a model for human Alport syndrome. Genetic ablation of either MMP-2 or MMP-9, or both MMP-2 and MMP-9, led to compensatory up-regulation of other MMPs in the kidney glomerulus. Pharmacological ablation of enzymatic activity associated with multiple GBM-degrading MMPs, before the onset of proteinuria or GBM structural defects in the alpha3(IV)-/- mice, led to significant attenuation in disease progression associated with delayed proteinuria and marked extension in survival. In contrast, inhibition of MMPs after induction of proteinuria led to acceleration of disease associated with extensive interstitial fibrosis and early death of alpha3(IV)-/- mice.
Conclusions: These results suggest that preserving GBM/extracellular matrix integrity before the onset of proteinuria leads to significant disease protection, but if this window of opportunity is lost, MMP-inhibition at the later stages of Alport disease leads to accelerated glomerular and interstitial fibrosis. Our findings identify a crucial dual role for MMPs in the progression of Alport disease in alpha3(IV)-/- mice, with an early pathogenic function and a later protective action. Hence, we propose possible use of MMP-inhibitors as disease-preventive drugs for patients with Alport syndrome with identified genetic defects, before the onset of proteinuria.
Conflict of interest statement
Figures
References
-
- Kalluri R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3:422–433. - PubMed
-
- Timpl R. Structure and biological activity of basement membrane proteins. Eur J Biochem. 1989;180:487–502. - PubMed
-
- Yurchenco PD. Assembly of basement membranes. Ann N Y Acad Sci. 1990;580:195–213. - PubMed
-
- Timpl R, Wiedemann H, van Delden V, Furthmayr H, Kuhn K. A network model for the organization of type IV collagen molecules in basement membranes. Eur J Biochem. 1981;120:203–211. - PubMed
-
- Longo I, Porcedda P, Mari F, Giachino D, Meloni I, et al. COL4A3/COL4A4 mutations: From familial hematuria to autosomal-dominant or recessive Alport syndrome. Kidney Int. 2002;61:1947–1956. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
