

A family of E. coli expression vectors for laboratory scale and high throughput soluble protein production
- PMID: 16509985
- PMCID: PMC1420288
- DOI: 10.1186/1472-6750-6-12
A family of E. coli expression vectors for laboratory scale and high throughput soluble protein production
Abstract
Background: In the past few years, both automated and manual high-throughput protein expression and purification has become an accessible means to rapidly screen and produce soluble proteins for structural and functional studies. However, many of the commercial vectors encoding different solubility tags require different cloning and purification steps for each vector, considerably slowing down expression screening. We have developed a set of E. coli expression vectors with different solubility tags that allow for parallel cloning from a single PCR product and can be purified using the same protocol.
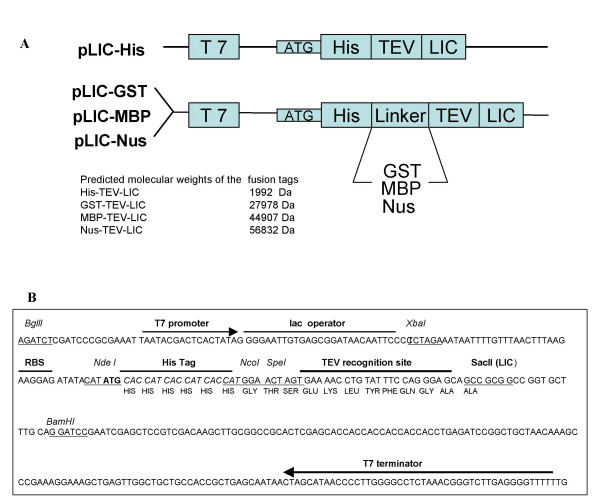
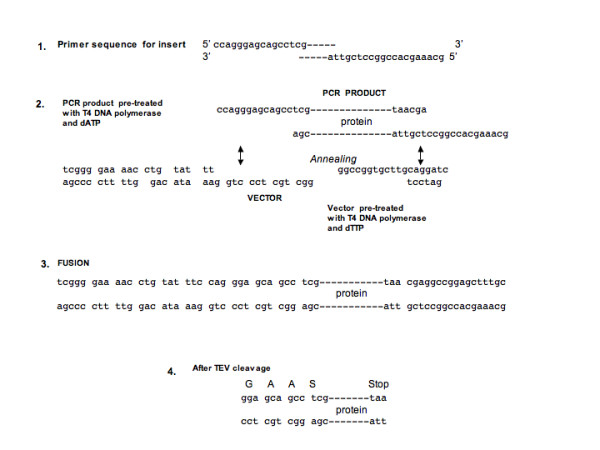
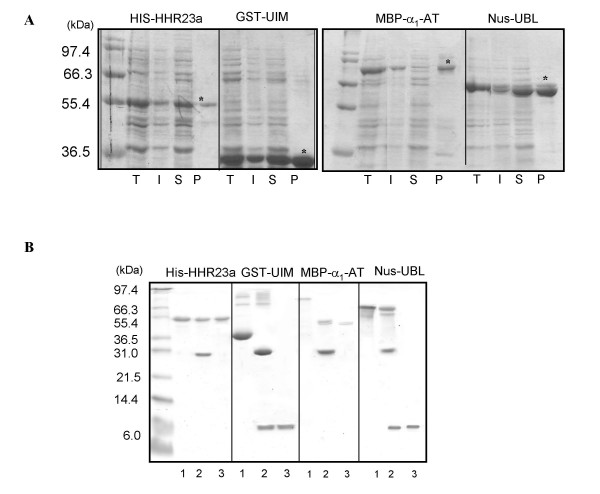
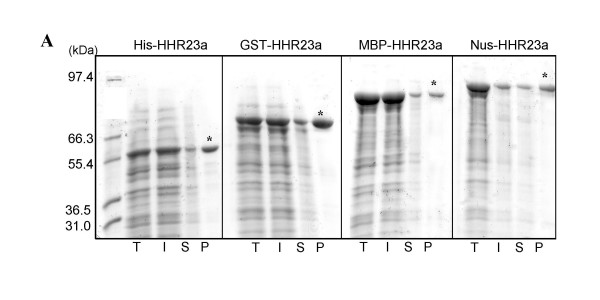
Results: The set of E. coli expression vectors, encode for either a hexa-histidine tag or the three most commonly used solubility tags (GST, MBP, NusA) and all with an N-terminal hexa-histidine sequence. The result is two-fold: the His-tag facilitates purification by immobilised metal affinity chromatography, whilst the fusion domains act primarily as solubility aids during expression, in addition to providing an optional purification step. We have also incorporated a TEV recognition sequence following the solubility tag domain, which allows for highly specific cleavage (using TEV protease) of the fusion protein to yield native protein. These vectors are also designed for ligation-independent cloning and they possess a high-level expressing T7 promoter, which is suitable for auto-induction. To validate our vector system, we have cloned four different genes and also one gene into all four vectors and used small-scale expression and purification techniques. We demonstrate that the vectors are capable of high levels of expression and that efficient screening of new proteins can be readily achieved at the laboratory level.
Conclusion: The result is a set of four rationally designed vectors, which can be used for streamlined cloning, expression and purification of target proteins in the laboratory and have the potential for being adaptable to a high-throughput screening.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
