

Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling
- PMID: 16511602
- PMCID: PMC1386110
- DOI: 10.1172/JCI27374
Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling
Abstract
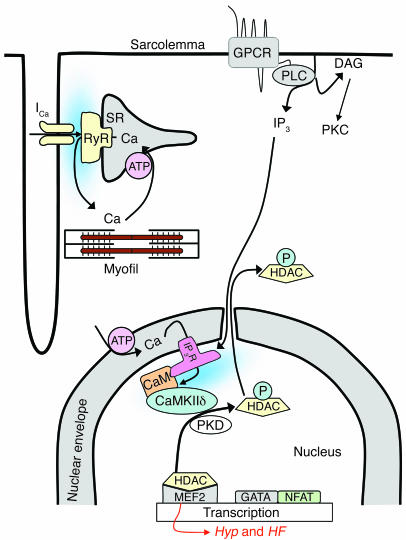
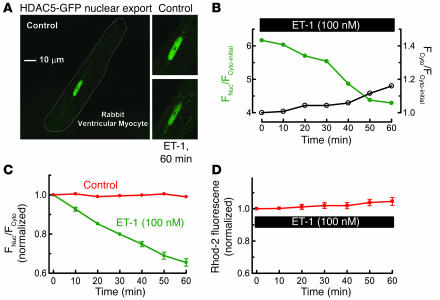
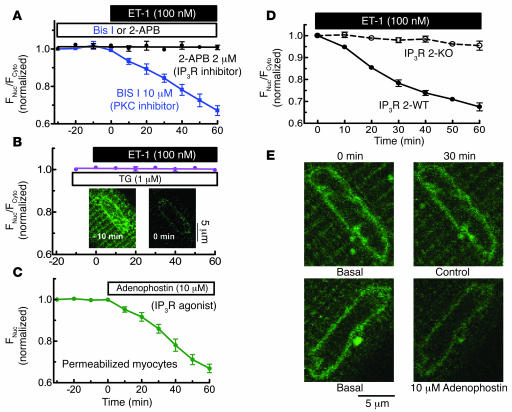
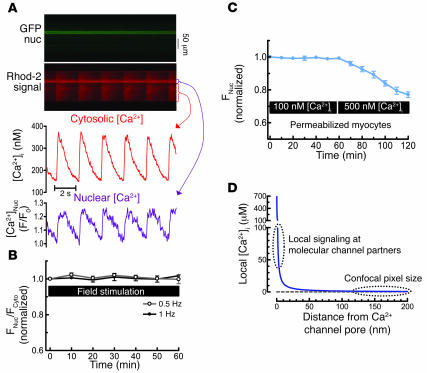
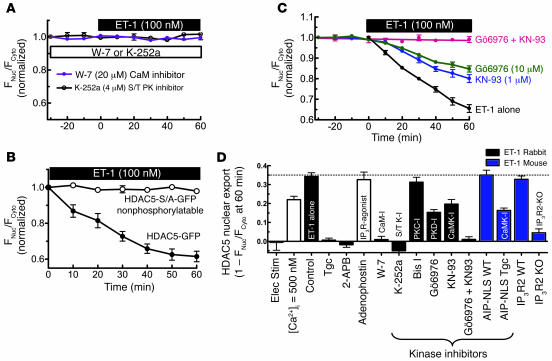
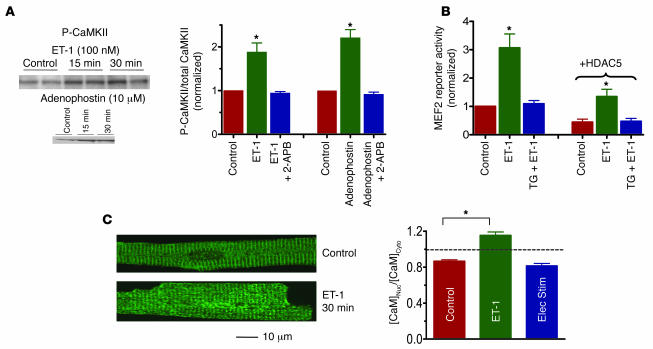
Previous work showed that calmodulin (CaM) and Ca2+-CaM-dependent protein kinase II (CaMKII) are somehow involved in cardiac hypertrophic signaling, that inositol 1,4,5-trisphosphate receptors (InsP3Rs) in ventricular myocytes are mainly in the nuclear envelope, where they associate with CaMKII, and that class II histone deacetylases (e.g., HDAC5) suppress hypertrophic gene transcription. Furthermore, HDAC phosphorylation in response to neurohumoral stimuli that induce hypertrophy, such as endothelin-1 (ET-1), activates HDAC nuclear export, thereby regulating cardiac myocyte transcription. Here we demonstrate a detailed mechanistic convergence of these 3 issues in adult ventricular myocytes. We show that ET-1, which activates plasmalemmal G protein-coupled receptors and InsP3 production, elicits local nuclear envelope Ca2+ release via InsP3R. This local Ca2+ release activates nuclear CaMKII, which triggers HDAC5 phosphorylation and nuclear export (derepressing transcription). Remarkably, this Ca2+-dependent pathway cannot be activated by the global Ca2+ transients that cause contraction at each heartbeat. This novel local Ca2+ signaling in excitation-transcription coupling is analogous to but separate (and insulated) from that involved in excitation-contraction coupling. Thus, myocytes can distinguish simultaneous local and global Ca2+ signals involved in contractile activation from those targeting gene expression.
Figures
Comment in
-
Dichotomy of Ca2+ in the heart: contraction versus intracellular signaling.J Clin Invest. 2006 Mar;116(3):623-6. doi: 10.1172/JCI27824. J Clin Invest. 2006. PMID: 16511595 Free PMC article. Review.
References
-
- Frey N, McKinsey TA, Olson EN. Decoding calcium signals involved in cardiac growth and function. Nat. Med. 2000;6:1221–1227. - PubMed
-
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109:S67–S79. - PubMed
-
- Zhu W, et al. Ca2+/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J. Biol. Chem. 2000;275:15239–15245. - PubMed
-
- Gruver CL, DeMayo F, Goldstein MA, Means AR. Targeted developmental overexpression of calmodulin induces proliferative and hypertrophic growth of cardiomyocytes in transgenic mice. Endocrinology. 1993;133:376–388. - PubMed
-
- Colomer JM, Means AR. Chronic elevation of calmodulin in the ventricles of transgenic mice increases the autonomous activity of calmodulin-dependent protein kinase II, which regulates atrial natriuretic factor gene expression. Mol. Endocrinol. 2000;14:1125–1136. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
