

Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action
- PMID: 16520744
- PMCID: PMC1617037
- DOI: 10.1038/sj.bjp.0706709
Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action
Abstract
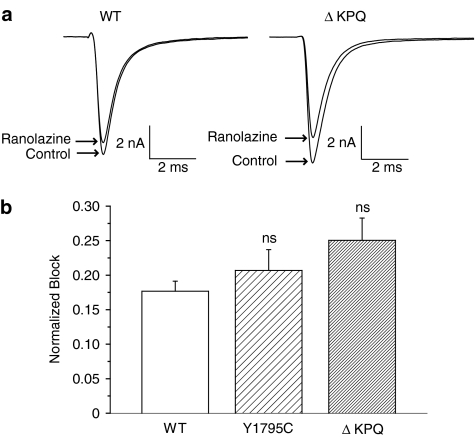
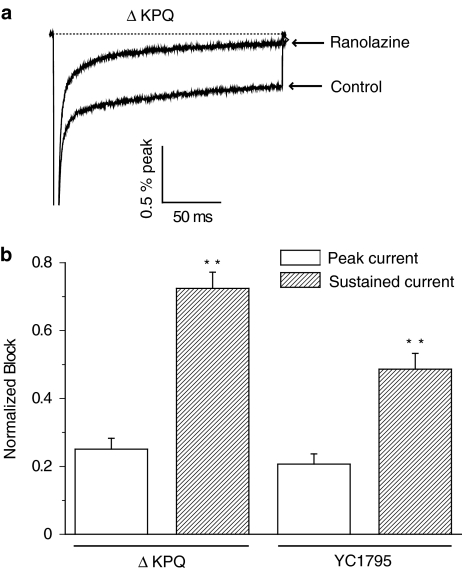
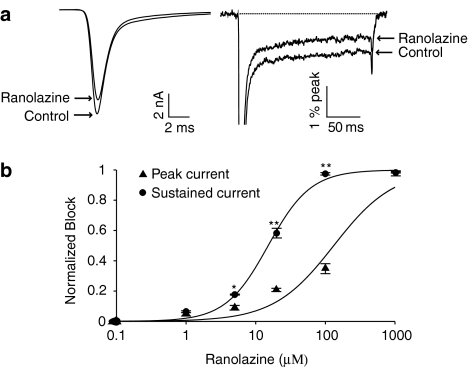
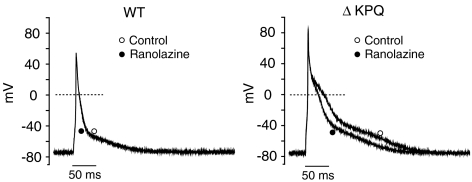
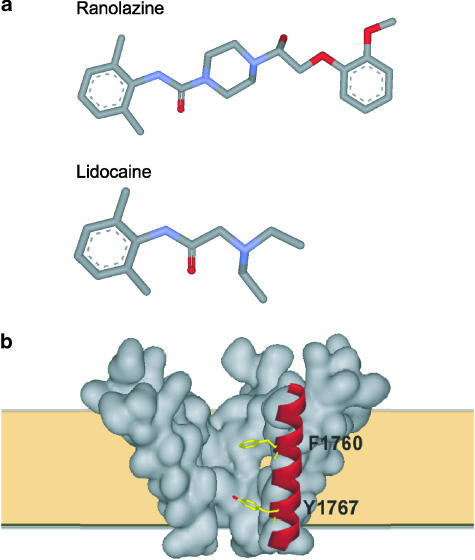
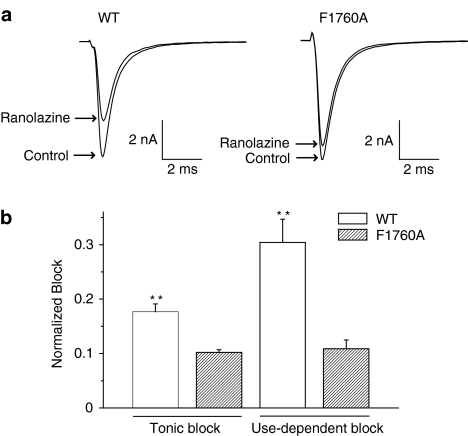
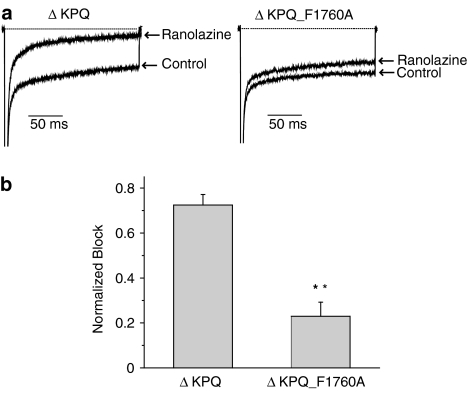
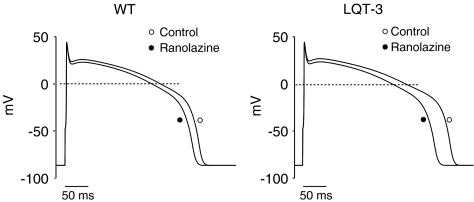
1 We studied the effects of ranolazine, an antianginal agent with promise as an antiarrhythmic drug, on wild-type (WT) and long QT syndrome variant 3 (LQT-3) mutant Na(+) channels expressed in human embryonic kidney (HEK) 293 cells and knock-in mouse cardiomyocytes and used site-directed mutagenesis to probe the site of action of the drug. 2 We find preferential ranolazine block of sustained vs peak Na(+) channel current for LQT-3 mutant (DeltaKPQ and Y1795C) channels (IC(50)=15 vs 135 microM) with similar results obtained in HEK 293 cells and knock-in myocytes. 3 Ranolazine block of both peak and sustained Na(+) channel current is significantly reduced by mutation (F1760A) of a single residue previously shown to contribute critically to the binding site for local anesthetic (LA) molecules in the Na(+) channel. 4 Ranolazine significantly decreases action potential duration (APD) at 50 and 90% repolarization by 23+/-5 and 27+/-3%, respectively, in DeltaKPQ mouse ventricular myocytes but has little effect on APD of WT myocytes. 5 Computational modeling of human cardiac myocyte electrical activity that incorporates our voltage-clamp data predicts marked ranolazine-induced APD shortening in cells expressing LQT-3 mutant channels. 6 Our results demonstrate for the first time the utility of ranolazine as a blocker of sustained Na(+) channel activity induced by inherited mutations that cause human disease and further, that these effects are very likely due to interactions of ranolazine with the receptor site for LA molecules in the sodium channel.
Figures
Comment in
-
Ranolazine and late cardiac sodium current--a therapeutic target for angina, arrhythmia and more?Br J Pharmacol. 2006 May;148(1):4-6. doi: 10.1038/sj.bjp.0706713. Br J Pharmacol. 2006. PMID: 16520741 Free PMC article.
Similar articles
-
Ranolazine and late cardiac sodium current--a therapeutic target for angina, arrhythmia and more?Br J Pharmacol. 2006 May;148(1):4-6. doi: 10.1038/sj.bjp.0706713. Br J Pharmacol. 2006. PMID: 16520741 Free PMC article.
-
State- and use-dependent block of muscle Nav1.4 and neuronal Nav1.7 voltage-gated Na+ channel isoforms by ranolazine.Mol Pharmacol. 2008 Mar;73(3):940-8. doi: 10.1124/mol.107.041541. Epub 2007 Dec 13. Mol Pharmacol. 2008. PMID: 18079277 Free PMC article.
-
Ranolazine for congenital and acquired late INa-linked arrhythmias: in silico pharmacological screening.Circ Res. 2013 Sep 13;113(7):e50-e61. doi: 10.1161/CIRCRESAHA.113.301971. Epub 2013 Jul 29. Circ Res. 2013. PMID: 23897695 Free PMC article.
-
Potential application of late sodium current blockade in the treatment of heart failure and atrial fibrillation.Rev Cardiovasc Med. 2009;10 Suppl 1:S46-52. Rev Cardiovasc Med. 2009. PMID: 19898288 Review.
-
Electrophysiologic properties and antiarrhythmic actions of a novel antianginal agent.J Cardiovasc Pharmacol Ther. 2004 Sep;9 Suppl 1:S65-83. doi: 10.1177/107424840400900106. J Cardiovasc Pharmacol Ther. 2004. PMID: 15378132 Review.
Cited by
-
Intracellular calcium attenuates late current conducted by mutant human cardiac sodium channels.Circ Arrhythm Electrophysiol. 2015 Aug;8(4):933-41. doi: 10.1161/CIRCEP.115.002760. Epub 2015 May 28. Circ Arrhythm Electrophysiol. 2015. PMID: 26022185 Free PMC article.
-
The citrus flavanone hesperetin preferentially inhibits slow-inactivating currents of a long QT syndrome type 3 syndrome Na+ channel mutation.Br J Pharmacol. 2019 Apr;176(8):1090-1105. doi: 10.1111/bph.14577. Epub 2019 Mar 27. Br J Pharmacol. 2019. PMID: 30650182 Free PMC article.
-
Glycolytic inhibition causes spontaneous ventricular fibrillation in aged hearts.Am J Physiol Heart Circ Physiol. 2011 Jul;301(1):H180-91. doi: 10.1152/ajpheart.00128.2011. Epub 2011 Apr 8. Am J Physiol Heart Circ Physiol. 2011. PMID: 21478408 Free PMC article.
-
Safety and Efficacy of Ranolazine for the Treatment of Chronic Angina Pectoris.Clin Med Insights Ther. 2013 Jan 15;2013(5):1-14. doi: 10.4137/CMT.S7824. Clin Med Insights Ther. 2013. PMID: 24574825 Free PMC article.
-
Differential Effect of Three Macrolide Antibiotics on Cardiac Pathology and Electrophysiology in a Myocardial Infarction Rat Model: Influence on Sodium Nav1.5 Channel Expression.Pharmaceuticals (Basel). 2021 Jun 22;14(7):597. doi: 10.3390/ph14070597. Pharmaceuticals (Basel). 2021. PMID: 34206182 Free PMC article.
References
-
- ABRIEL H., CABO C., WEHRENS X.H., RIVOLTA I., MOTOIKE H.K., MEMMI M., NAPOLITANO C., PRIORI S.G., KASS R.S. Novel arrhythmogenic mechanism revealed by a long-QT syndrome mutation in the cardiac Na(+) channel. Circ. Res. 2001;88:740–745. - PubMed
-
- ABRIEL H., WEHRENS X.H., BENHORIN J., KEREM B., KASS R.S. Molecular pharmacology of the sodium channel mutation D1790G linked to the long-QT syndrome. Circulation. 2000;102:921–925. - PubMed
-
- AN R.H., BANGALORE R., ROSERO S.Z., KASS R.S. Lidocaine block of LQT-3 mutant human Na+ channels. Circ. Res. 1996;79:103–108. - PubMed
-
- AN R.H., WANG X.L., KEREM B., BENHORIN J., MEDINA A., GOLDMIT M., KASS R.S. Novel LQT-3 mutation affects Na+ channel activity through interactions between alpha- and beta1-subunits. Circ. Res. 1998;83:141–146. - PubMed
-
- ANTZELEVITCH C., BELARDINELLI L., WU L., FRASER H., ZYGMUNT A.C., BURASHNIKOV A., DIEGO J.M., FISH J.M., CORDEIRO J.M., GOODROW R.J., Jr, SCORNIK F., PEREZ G. Electrophysiologic properties and antiarrhythmic actions of a novel antianginal agent. J. Cardiovasc. Pharmacol. Ther. 2004a;9 Suppl 1:S65–S83. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
