

A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase
- PMID: 16532393
- PMCID: PMC1424677
- DOI: 10.1086/502802
A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase
Abstract
We present a statistical model for patterns of genetic variation in samples of unrelated individuals from natural populations. This model is based on the idea that, over short regions, haplotypes in a population tend to cluster into groups of similar haplotypes. To capture the fact that, because of recombination, this clustering tends to be local in nature, our model allows cluster memberships to change continuously along the chromosome according to a hidden Markov model. This approach is flexible, allowing for both "block-like" patterns of linkage disequilibrium (LD) and gradual decline in LD with distance. The resulting model is also fast and, as a result, is practicable for large data sets (e.g., thousands of individuals typed at hundreds of thousands of markers). We illustrate the utility of the model by applying it to dense single-nucleotide-polymorphism genotype data for the tasks of imputing missing genotypes and estimating haplotypic phase. For imputing missing genotypes, methods based on this model are as accurate or more accurate than existing methods. For haplotype estimation, the point estimates are slightly less accurate than those from the best existing methods (e.g., for unrelated Centre d'Etude du Polymorphisme Humain individuals from the HapMap project, switch error was 0.055 for our method vs. 0.051 for PHASE) but require a small fraction of the computational cost. In addition, we demonstrate that the model accurately reflects uncertainty in its estimates, in that probabilities computed using the model are approximately well calibrated. The methods described in this article are implemented in a software package, fastPHASE, which is available from the Stephens Lab Web site.
Figures
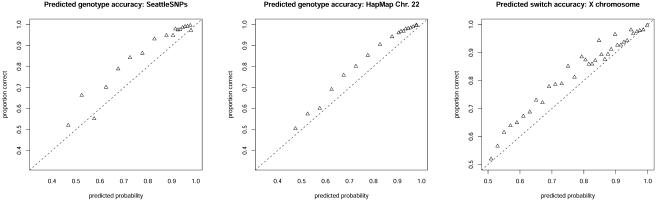
References
Web Resources
-
- HAP Web site, http://research.calit2.net/hap/
-
- HaploBlock, http://bioinfo.cs.technion.ac.il/haploblock/
-
- International HapMap Project, http://www.hapmap.org/
-
- SeattleSNPs, http://pga.gs.washington.edu
References
-
- Akaike H (1974) A new look at the statistical model identification. IEEE Trans Automatic Control AC 19:719–723
-
- Bates JM, Granger CWJ (1969) The combination of forecasts. Oper Res Q 20:451–468
-
- Breiman L (1996) Bagging predictors. Mach Learn 24:123–140
-
- Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Ser B 39:1–38
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
