

Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity
- PMID: 16547140
- PMCID: PMC1458799
- DOI: 10.1073/pnas.0508156103
Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity
Abstract
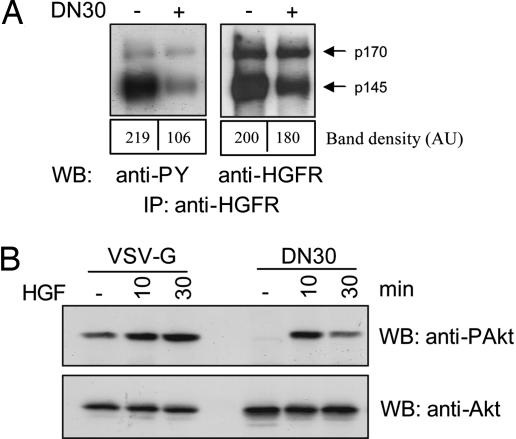
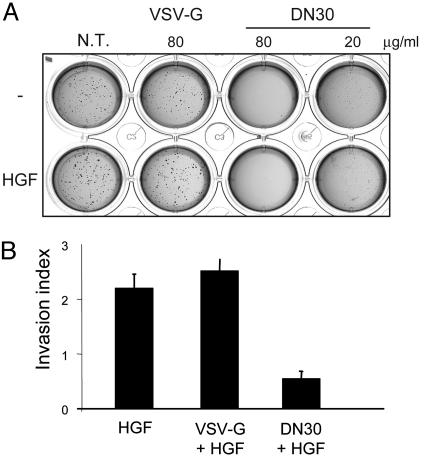
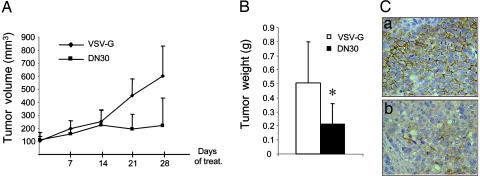
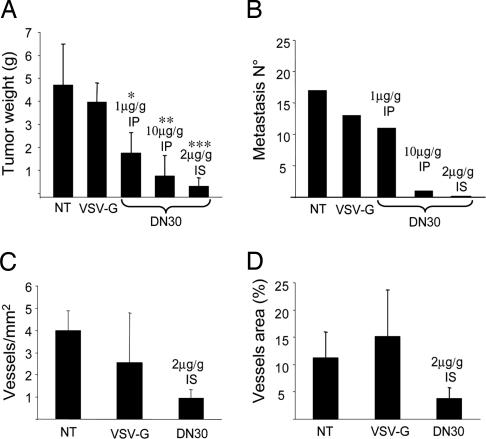
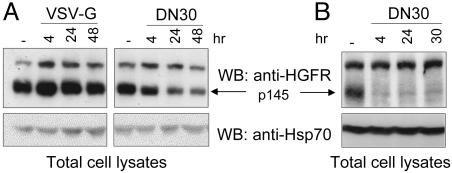
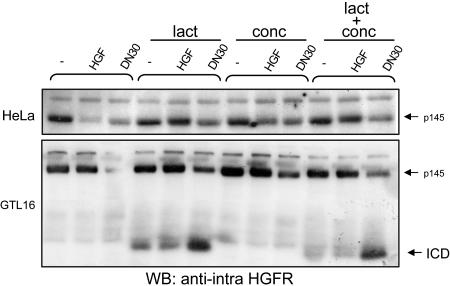
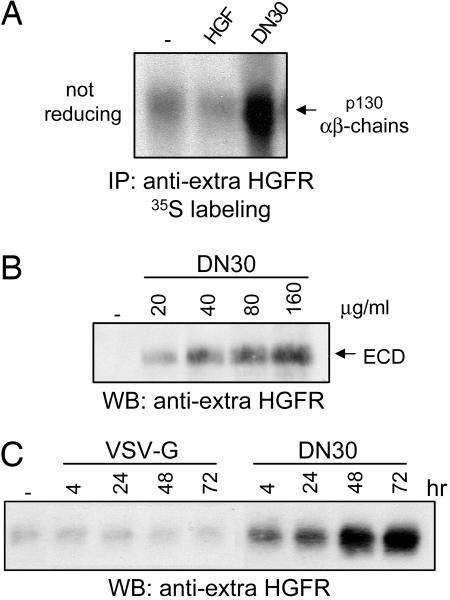
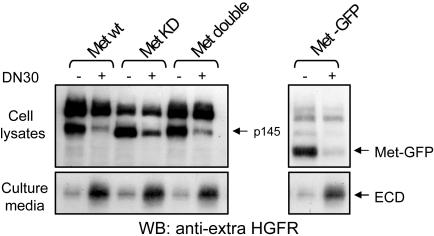
Targeting tyrosine kinase receptors (RTKs) with specific Abs is a promising therapeutic approach for cancer treatment, although the molecular mechanism(s) responsible for the Abs' biological activity are not completely known. We targeted the transmembrane RTK for hepatocyte growth factor (HGF) with a monoclonal Ab (DN30). In vitro, chronic treatment of carcinoma cell lines resulted in impairment of HGF-induced signal transduction, anchorage-independent growth, and invasiveness. In vivo, administration of DN30 inhibited growth and metastatic spread to the lung of neoplastic cells s.c. transplanted into immunodeficient nu/nu mice. This Ab efficiently down-regulates HGF receptor through a molecular mechanism involving a double proteolytic cleavage: (i) cleavage of the extracellular portion, resulting in "shedding" of the ectodomain, and (ii) cleavage of the intracellular domain, which is rapidly degraded by the proteasome. Interestingly, the "decoy effect" generated by the shed ectodomain, acting as a dominant negative molecule, enhanced the inhibitory effect of the Ab.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures
Similar articles
-
Deletion of the ectodomain unleashes the transforming, invasive, and tumorigenic potential of the MET oncogene.Cancer Sci. 2009 Apr;100(4):633-8. doi: 10.1111/j.1349-7006.2008.01079.x. Epub 2009 Jan 21. Cancer Sci. 2009. PMID: 19175607 Free PMC article.
-
Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody.J Biol Chem. 2010 Nov 12;285(46):36149-57. doi: 10.1074/jbc.M110.134031. Epub 2010 Sep 10. J Biol Chem. 2010. PMID: 20833723 Free PMC article.
-
c-Met ectodomain shedding rate correlates with malignant potential.Clin Cancer Res. 2006 Jul 15;12(14 Pt 1):4154-62. doi: 10.1158/1078-0432.CCR-06-0250. Clin Cancer Res. 2006. PMID: 16857786
-
Structure, biosynthesis and biochemical properties of the HGF receptor in normal and malignant cells.EXS. 1993;65:131-65. EXS. 1993. PMID: 8380735 Review.
-
State of the structure address on MET receptor activation by HGF.Biochem Soc Trans. 2021 Apr 30;49(2):645-661. doi: 10.1042/BST20200394. Biochem Soc Trans. 2021. PMID: 33860789 Free PMC article. Review.
Cited by
-
Inhibition of ligand-independent constitutive activation of the Met oncogenic receptor by the engineered chemically-modified antibody DN30.Mol Oncol. 2015 Nov;9(9):1760-72. doi: 10.1016/j.molonc.2015.05.007. Epub 2015 Jun 5. Mol Oncol. 2015. PMID: 26119717 Free PMC article.
-
The MET Oncogene as a Therapeutical Target in Cancer Invasive Growth.Front Pharmacol. 2012 Sep 11;3:164. doi: 10.3389/fphar.2012.00164. eCollection 2012. Front Pharmacol. 2012. PMID: 22973229 Free PMC article.
-
Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases.FEBS J. 2015 Oct;282(19):3693-721. doi: 10.1111/febs.13342. Epub 2015 Jul 4. FEBS J. 2015. PMID: 26096795 Free PMC article. Review.
-
c-MET: an exciting new target for anticancer therapy.Ther Adv Med Oncol. 2011 Nov;3(1 Suppl):S3-5. doi: 10.1177/1758834011423402. Ther Adv Med Oncol. 2011. PMID: 22128286 Free PMC article. No abstract available.
-
Proteolytic cleavages of MET: the divide-and-conquer strategy of a receptor tyrosine kinase.BMB Rep. 2019 Apr;52(4):239-249. doi: 10.5483/BMBRep.2019.52.4.024. BMB Rep. 2019. PMID: 30670153 Free PMC article. Review.
References
-
- Hudson P. J. Curr. Opin. Immunol. 1999;11:548–557. - PubMed
-
- Hudson P. J., Souriau C. Nat. Med. 2003;9:129–134. - PubMed
-
- Gschwind A., Fischer O. M., Ullrich A. Nat. Rev. Cancer. 2004;4:361–370. - PubMed
-
- Cragg M. S., French R. R., Glennie M. J. Curr. Opin. Immunol. 1999;11:541–547. - PubMed
-
- Ferrara N., Hillan K. J., Gerber H. P., Novotny W. Nat. Rev. Drug Discovery. 2004;3:391–400. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
