

Spectral clustering of protein sequences
- PMID: 16547200
- PMCID: PMC1409676
- DOI: 10.1093/nar/gkj515
Spectral clustering of protein sequences
Abstract
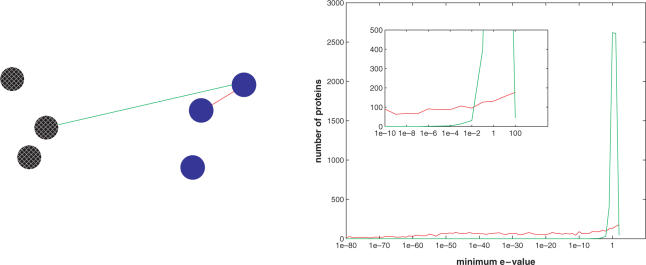
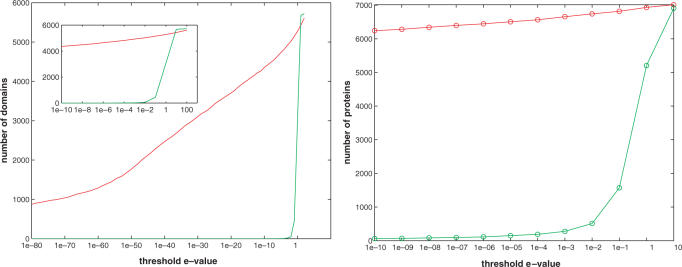
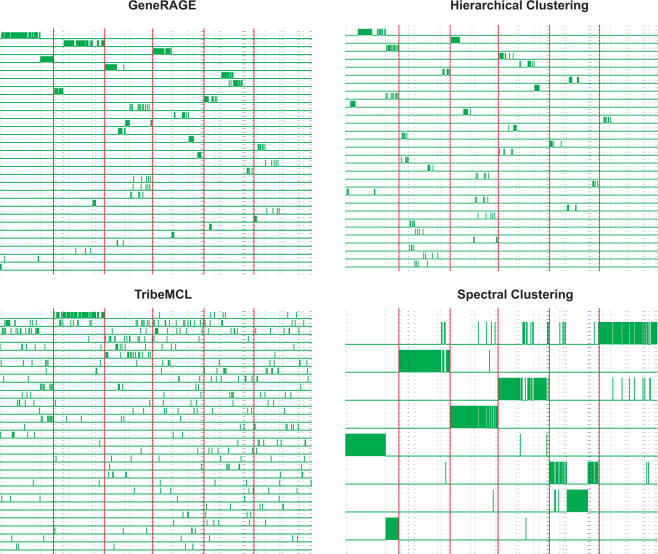
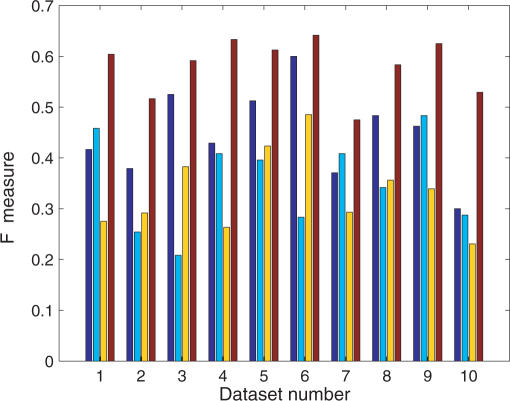
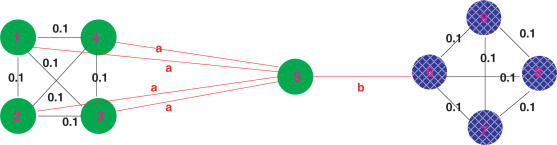
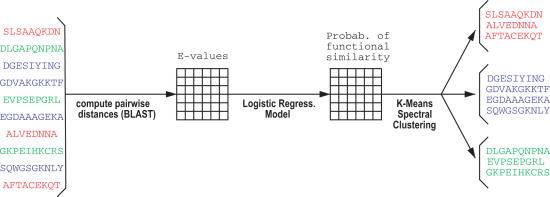
An important problem in genomics is automatically clustering homologous proteins when only sequence information is available. Most methods for clustering proteins are local, and are based on simply thresholding a measure related to sequence distance. We first show how locality limits the performance of such methods by analysing the distribution of distances between protein sequences. We then present a global method based on spectral clustering and provide theoretical justification of why it will have a remarkable improvement over local methods. We extensively tested our method and compared its performance with other local methods on several subsets of the SCOP (Structural Classification of Proteins) database, a gold standard for protein structure classification. We consistently observed that, the number of clusters that we obtain for a given set of proteins is close to the number of superfamilies in that set; there are fewer singletons; and the method correctly groups most remote homologs. In our experiments, the quality of the clusters as quantified by a measure that combines sensitivity and specificity was consistently better [on average, improvements were 84% over hierarchical clustering, 34% over Connected Component Analysis (CCA) (similar to GeneRAGE) and 72% over another global method, TribeMCL].
Figures
References
-
- Ballard D., Brown C. Computer Vision. Englewood Cliffs: Prentice-Hall; 1982.
-
- Enright A.J., Ouzounis C.A. GeneRAGE: a robust algorithm for sequence clustering and domain detection. Bioinformatics. 2000;16:451–457. - PubMed
-
- Pipenbacher P., Schliep A., Schneckener S., Schonhuth A., Schomburg D., Schrader R. ProClust: improved clustering of protein sequences with an extended graph-based approach. Bioinformatics. 2002;18:S182–S191. - PubMed
