

Review
doi: 10.1038/nature04712.
The ABC protein turned chloride channel whose failure causes cystic fibrosis
Affiliations
- PMID: 16554808
- PMCID: PMC2720541
- DOI: 10.1038/nature04712
Item in Clipboard
Review
The ABC protein turned chloride channel whose failure causes cystic fibrosis
Nature.
.
Abstract
CFTR chloride channels are encoded by the gene mutated in patients with cystic fibrosis. These channels belong to the superfamily of ABC transporter ATPases. ATP-driven conformational changes, which in other ABC proteins fuel uphill substrate transport across cellular membranes, in CFTR open and close a gate to allow transmembrane flow of anions down their electrochemical gradient. New structural and biochemical information from prokaryotic ABC proteins and functional information from CFTR channels has led to a unifying mechanism explaining those ATP-driven conformational changes.
Figures
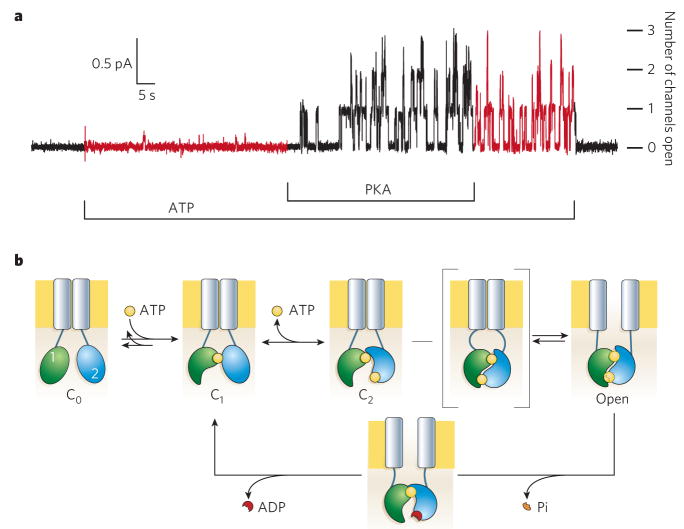
a, CFTR’s regulatory (R) domain must be phosphorylated by PKA before ATP is able to support channel opening. The recording shows chloride current flow through individual CFTR channels upon opening. Endogenous membrane-attached phosphatases partly dephosphorylate the R domain on PKA withdrawal, reducing the probability of finding a channel open. But channels do not stop opening until ATP is removed; numbers of simultaneously open channels are indicated at the right. b, Present structural interpretation of ATP-dependent gating cycle of phosphorylated CFTR channels. The R domain is omitted. ATP (yellow) remains tightly bound to NBD1 (green) Walker motifs for several minutes, during which time many closed–open–closed gating cycles occur. ATP binding to NBD2 (blue) is followed by a slow channel opening step (C2-to-Open) that proceeds through a transition state (square brackets) in which the intramolecular NBD1–NBD2 tight heterodimer is formed but the transmembrane pore (grey rectangles) has not yet opened. The relatively stable open state becomes destabilized by hydrolysis of the ATP bound at the NBD2 composite catalytic site and loss of the hydrolysis product, inorganic phosphate (Pi). The ensuing disruption of the tight dimer interface leads to channel closure.
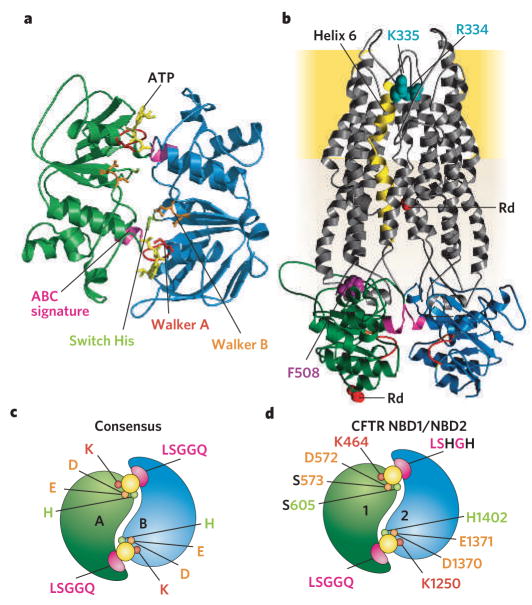
a, Homology model of CFTR NBD heterodimer (NBD1 in green, NBD2 in blue) based on several ATP (yellow) bound NBD crystals, including human CFTR NBD1 F508A and dimeric MJ0796, MalK and HlyB. b, Highly speculative homology model of CFTR including its two TMDs (grey) modelled on bacterial MsbA structures. Each TMD comprises cytoplasmic segments that link membrane-embedded α-helices (one directly) to the NBDs (colour-coded as in a). Membrane-spanning helix 6 (gold) has been implicated in forming part of the anion-permeation pathway, and positively charged residues (blue-green) in it are thought to recruit anions into an outer vestibule. In CF patients, deletion of Phe 508 (purple), at the interface between NBD1 and TMD1 causes post-translational misfolding and degradation. c, d, Illustrations contrasting NBD-homodimer (c; for example, MJo796 and MalK) consensus catalytic site residues (for example, MJ0796 aand MalK) and expected CFTR NBD1–NBD2 heterodimer (d) with only one consensus catalytic site.
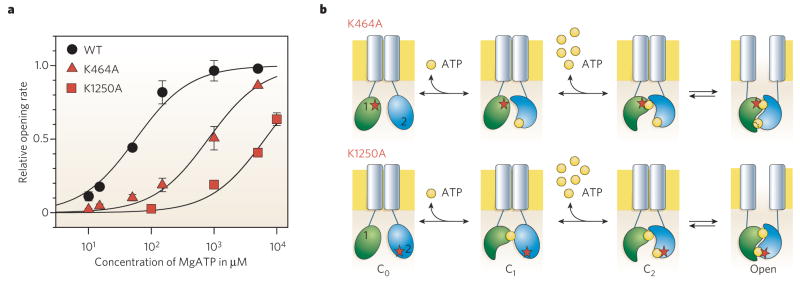
a, Dependence on cytoplasmic-surface ATP concentration of rate of channel opening for wild-type CFTR (WT) and for CFTR channels with a single mutation of the NBD1 (K464) or NBD2 (K1250) Walker A lysine. Either mutation lowers the apparent affinity for ATP-dependent channel opening, implying that ATP normally binds at both sites before a channel opens. b, Diagram illustrating the simplest interpretation of results in a. At lower [ATP], channel opening is limited by low success rate of ATP binding to the composite site that harbours the mutation (red star) of the conserved lysine. ATP first binds to the non-mutant site, that is to the NBD2 site (blue) in K464A channels (upper row), but to the NDB1 site (green) in K1250A channels (lower row). When at higher concentrations, ATP also occupies the mutant site, the NBDs dimerize and the channel opens.
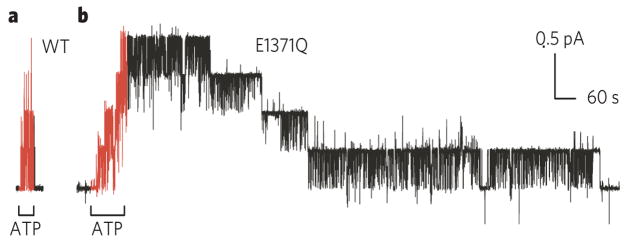
Comparison of speed of closing of wild-type CFTR (a) and of mutant CFTR bearing the single mutation E1371Q (b), of the conserved Walker B glutamate. This glutamate is required for ATP hydrolysis, but not for binding of ATP,,,. The greatly delayed closing of all four E1371Q channels (b), compared with the four WT CFTR channels (a), after removal of ATP supports the conclusion that ATP hydrolysis at the NBD2 composite catalytic site controls normal channel closing–,–,, and that the channel open-burst state corresponds to NBD dimerization,,,,.
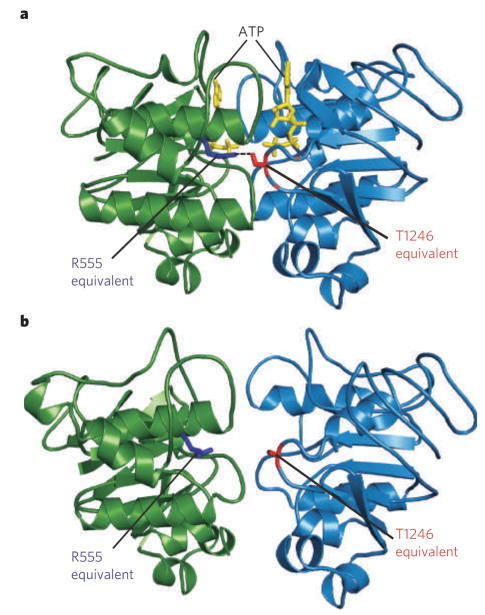
The MalK homodimer was crystallized with (a) and without (b) ATP (yellow). The structures illustrate pincer-like motion of MalK that closes the catalytic interface upon binding of two ATP molecules. The residues corresponding to CFTR R555 (blue) and T1246 (red) form a hydrogen bond (a, dotted line) linking Walker A of one subunit with the signature sequence of the other, only in the ATP-bound crystal (a).
References
-
- Rommens JM, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. - PubMed
-
- Riordan JR, et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Quinton PM. Chloride impermeability in cystic fibrosis. Nature. 1983;301:421–422. - PubMed
-
- Schoumacher RA, et al. Phosphorylation fails to activate chloride channels from cystic fibrosis airway cells. Nature. 1987;330:752–754. - PubMed
-
- Li M, et al. Cyclic AMP-dependent protein kinase opens chloride channels in normal but not cystic fibrosis airway epithelium. Nature. 1988;331:358–360. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
