

Essential role of Smad3 in angiotensin II-induced vascular fibrosis
- PMID: 16556868
- PMCID: PMC1450325
- DOI: 10.1161/01.RES.0000218782.52610.dc
Essential role of Smad3 in angiotensin II-induced vascular fibrosis
Abstract
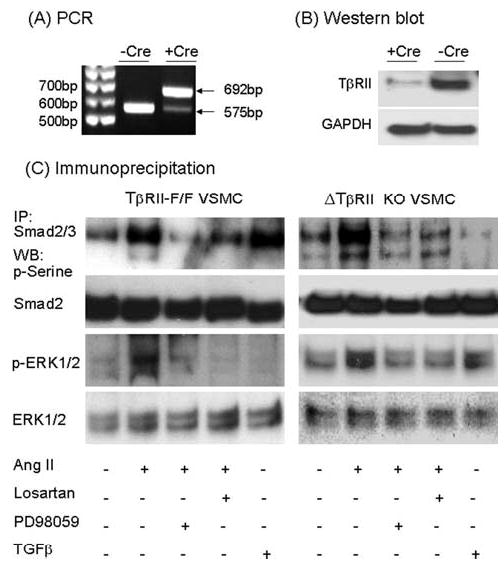
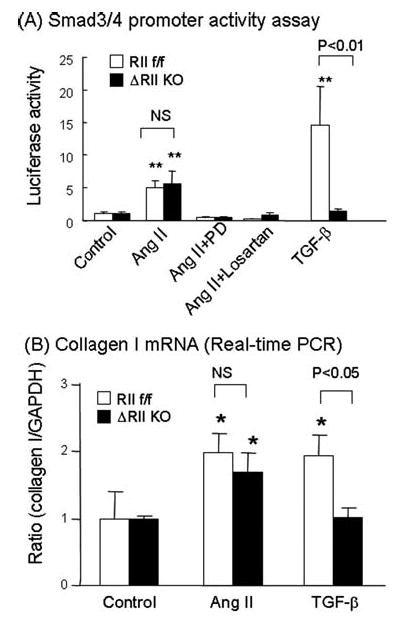
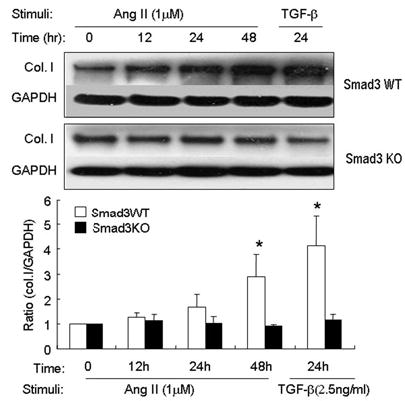
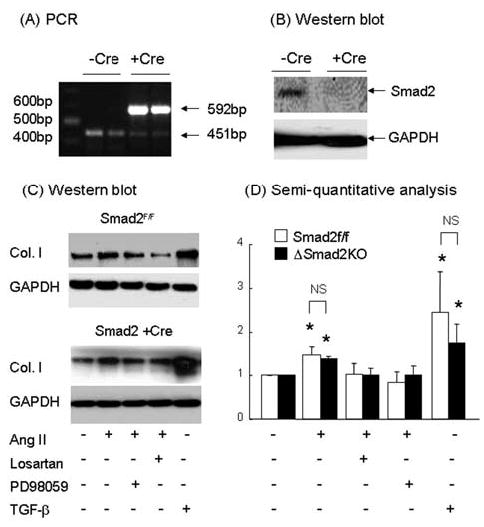
Angiotensin II (Ang II) plays a pivotal role in vascular fibrosis, which leads to serious complications in hypertension and diabetes. However, the underlying signaling mechanisms are largely unclear. In hypertensive patients, we found that arteriosclerosis was associated with the activation of Smad2/3. This observation was further investigated in vitro by stimulating mouse primary aorta vascular smooth muscle cells (VSMCs) with Ang II. There were several novel findings. First, Ang II was able to activate an early Smad signaling pathway directly at 15 to 30 minutes. This was extracellular signal-regulated kinase 1/2 (ERK1/2) mitogen-activated protein kinase (MAPK) dependent but transforming growth factor-beta (TGF-beta) independent because Ang II-induced Smad signaling was blocked by addition of ERK1/2 inhibitor and by dominant-negative (DN) ERK1/2 but not by DN-TGF-beta receptor II (TbetaRII) or conditional deletion of TbetaRII. Second, Ang II was also able to activate the late Smad2/3 signaling pathway at 24 hours, which was TGF-beta dependent because it was blocked by the anti-TGF-beta antibody and DN-TbetaRII. Finally, activation of Smad3 but not Smad2 was a key and necessary mechanism of Ang II-induced vascular fibrosis because Ang II induced Smad3/4 promoter activities and collagen matrix expression was abolished in VSMCs null for Smad3 but not Smad2. Thus, we concluded that Ang II induces vascular fibrosis via both TGF-beta-dependent and ERK1/2 MAPK-dependent Smad signaling pathways. Activation of Smad3 but not Smad2 is a key mechanism by which Ang II mediates arteriosclerosis.
Figures
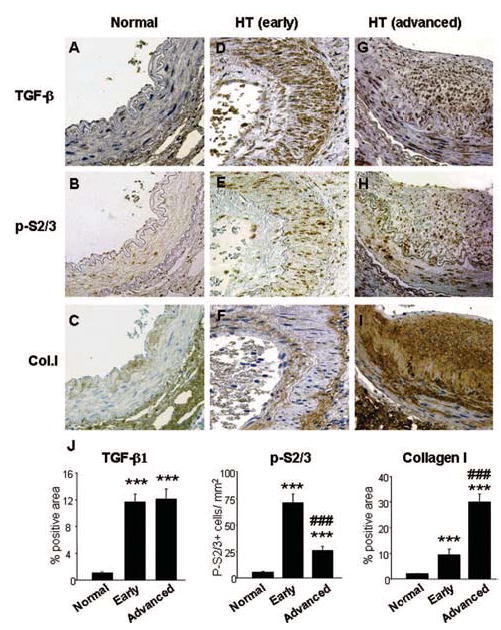
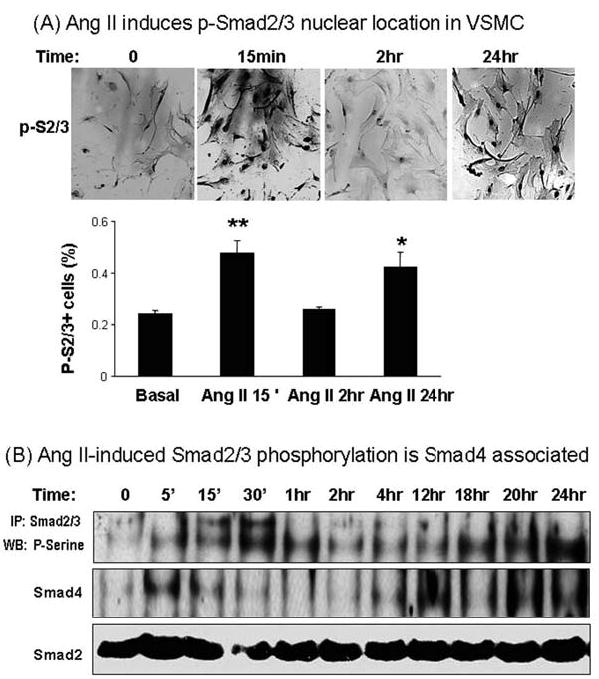
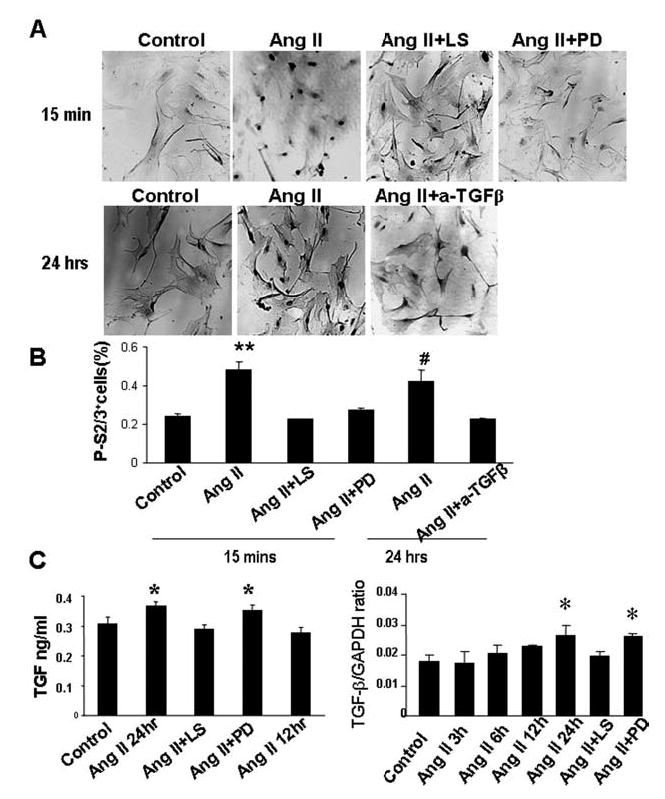
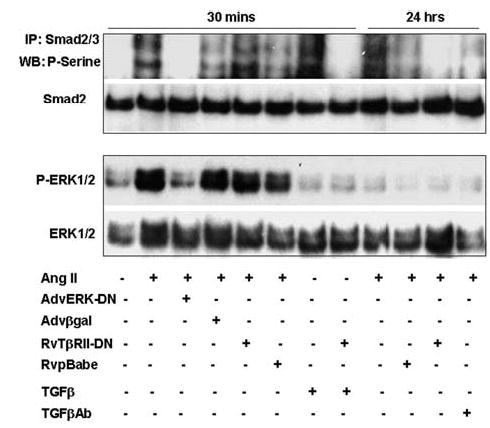
Comment in
-
Smad3 mediates angiotensin II- and TGF-beta1-induced vascular fibrosis: Smad3 thickens the plot.Circ Res. 2006 Apr 28;98(8):988-9. doi: 10.1161/01.RES.0000221824.87718.c0. Circ Res. 2006. PMID: 16645147 No abstract available.
References
-
- Ford CM, Li S, Pickering JG, Itoh H, Mukoyama M, Pratt RE, Gibbons GH, Dzau VJ. Angiotensin II stimulates collagen synthesis in human vascular smooth muscle cells. Involvement of the AT(1) receptor, transforming growth factor-beta, and tyrosine phosphorylation. Arterioscler Thromb Vasc Biol. 1999;19:1843–1851. - PubMed
-
- Miao CY, Tao X, Gong K, Zhang SH, Chu ZX, Su DF. Arterial remodeling in chronic sinoaortic-denervated rats. J Cardiovasc Pharmacol. 2001;37:6–15. - PubMed
-
- Lombardi DM, Viswanathan M, Vio CP, Saavedra JM, Schwartz SM, Johnson RJ. Renal and vascular injury induced by exogenous angiotensin II is AT1 receptor-dependent. Nephron. 2001;87:66–74. - PubMed
-
- Kakinuma Y, Kawamura T, Bills T, Yoshioka T, Ichikawa I, Fogo A. Blood pressure-independent effect of angiotensin inhibition on vascular lesions of chronic renal failure. Kidney Int. 1992;42:46–55. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
