

Etiology of insulin resistance
- PMID: 16563942
- PMCID: PMC2995525
- DOI: 10.1016/j.amjmed.2006.01.009
Etiology of insulin resistance
Abstract
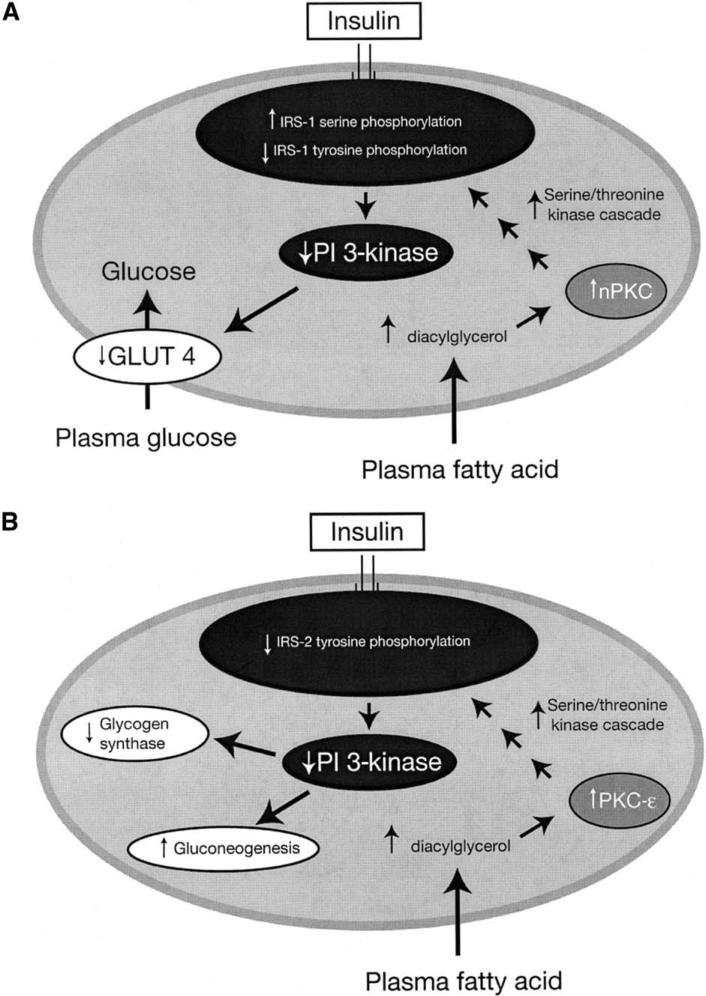
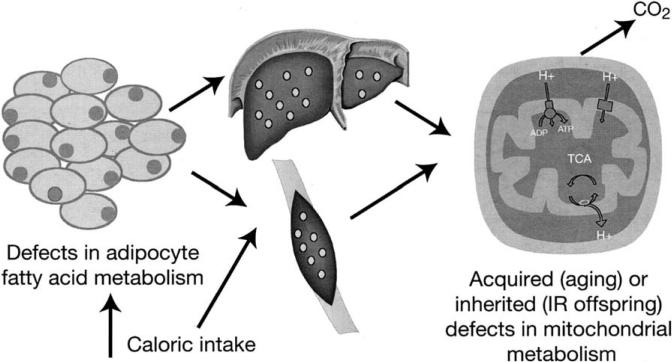
Type 2 diabetes mellitus is a major cause of morbidity and mortality worldwide, and the prevalence is set to increase dramatically over the coming decades. Understanding the metabolic pathways that lead to type 2 diabetes is therefore an important healthcare objective. Novel investigational techniques based on magnetic resonance spectroscopy (MRS) have allowed real-time insight into the molecular defects in patients with type 2 diabetes, revealing that insulin resistance is a product of decreased insulin-stimulated skeletal muscle glycogen synthesis, which can mostly be attributed to decreased insulin-stimulated glucose transport (Glut 4) activity. This defect appears to be a result of intracellular lipid-induced inhibition of insulin-stimulated insulin-receptor substrate (IRS)-1 tyrosine phosphorylation resulting in reduced IRS-1-associated phosphatidyl inositol 3 kinase activity. The hypothesis that insulin resistance is a result of accumulation of intracellular lipid metabolites (e.g., fatty acyl CoAs, diacylglycerol) in skeletal muscle and hepatocytes is supported by observations in patients and mouse models of lipodystrophy. Furthermore, the increase in hepatic insulin sensitivity observed in patients with type 2 diabetes following weight loss is also accompanied by a significant reduction in intrahepatic fat without any changes in circulating adipocytokines (interleukin-6, resistin, leptin). Finally, recent MRS studies in healthy, lean, elderly subjects and lean insulin-resistant offspring of parents with type 2 diabetes have demonstrated that reduced mitochondrial activity may also lead to increased intramyocellular lipid content and insulin resistance in skeletal muscle in these individuals. In summary, in vivo MRS has proved to be an important tool for elucidating the causal chain of events that causes insulin resistance. Understanding the cellular mechanism(s) of insulin resistance in turn offers the prospect of better targeted and more effective therapeutic interventions for treatment and prevention of type 2 diabetes.
Figures

References
-
- Williamson DF, Vinicor F, Bowman BA. Primary prevention of type 2 diabetes mellitus by lifestyle intervention: implications for health policy. Ann Intern Med. 2004;40:951–957. - PubMed
-
- US Renal Data System USRDS 2004 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. [May 26, 2005]. Available at: http://www.usrds.org/atlas_2004.htm.
-
- Ulbrecht JS, Cavanagh PR, Caputo GM. Foot problems in diabetes: an overview. Clin Infect Dis. 2004;39(suppl 2):S73–S82. - PubMed
-
- Hogan P, Dall T, Nikolov P. Economic costs of diabetes in the US in 2002. Diabetes Care. 2003;26:917–932. - PubMed
-
- Lucioni C, Garancini MP, Massi-Benedetti M, et al. The costs of type 2 diabetes mellitus in Italy: a CODE-2 substudy. Treat Endocrinol. 2003;2:121–133. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
