

A high-resolution map of transcription in the yeast genome
- PMID: 16569694
- PMCID: PMC1414796
- DOI: 10.1073/pnas.0601091103
A high-resolution map of transcription in the yeast genome
Abstract
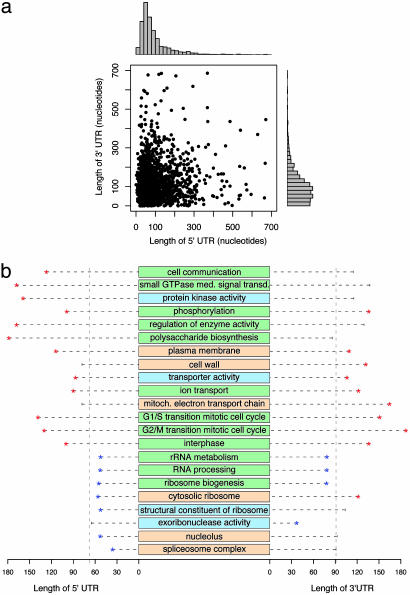
There is abundant transcription from eukaryotic genomes unaccounted for by protein coding genes. A high-resolution genome-wide survey of transcription in a well annotated genome will help relate transcriptional complexity to function. By quantifying RNA expression on both strands of the complete genome of Saccharomyces cerevisiae using a high-density oligonucleotide tiling array, this study identifies the boundary, structure, and level of coding and noncoding transcripts. A total of 85% of the genome is expressed in rich media. Apart from expected transcripts, we found operon-like transcripts, transcripts from neighboring genes not separated by intergenic regions, and genes with complex transcriptional architecture where different parts of the same gene are expressed at different levels. We mapped the positions of 3' and 5' UTRs of coding genes and identified hundreds of RNA transcripts distinct from annotated genes. These nonannotated transcripts, on average, have lower sequence conservation and lower rates of deletion phenotype than protein coding genes. Many other transcripts overlap known genes in antisense orientation, and for these pairs global correlations were discovered: UTR lengths correlated with gene function, localization, and requirements for regulation; antisense transcripts overlapped 3' UTRs more than 5' UTRs; UTRs with overlapping antisense tended to be longer; and the presence of antisense associated with gene function. These findings may suggest a regulatory role of antisense transcription in S. cerevisiae. Moreover, the data show that even this well studied genome has transcriptional complexity far beyond current annotation.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures



References
-
- Wilkie G. S., Dickson K. S., Gray N. K. Trends Biochem. Sci. 2003;28:182–188. - PubMed
-
- Storz G., Altuvia S., Wassarman K. M. Annu. Rev. Biochem. 2005;74:199–217. - PubMed
-
- Katayama S., Tomaru Y., Kasukawa T., Waki K., Nakanishi M., Nakamura M., Nishida H., Yap C. C., Suzuki M., Kawai J., et al. Science. 2005;309:1564–1566. - PubMed
-
- Mattick J. S. Nat. Rev. Genet. 2004;5:316–323. - PubMed
-
- Cheng J., Kapranov P., Drenkow J., Dike S., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G., et al. Science. 2005;308:1149–1154. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
