

Spontaneous rescue from cystic fibrosis in a mouse model
- PMID: 16571105
- PMCID: PMC1448185
- DOI: 10.1186/1471-2156-7-18
Spontaneous rescue from cystic fibrosis in a mouse model
Abstract
Background: From the original CftrTgH(neoim)Hgu mutant mouse model with a divergent genetic background (129P2, C57BL/6, MF1) we have generated two inbred CftrTgH(neoim)Hgu mutant strains named CF/1-CftrTgH(neoim)Hgu and CF/3-CftrTgH(neoim)Hgu, which are fertile and show normal growth and lifespan. Initial genome wide scan analysis with microsatellite markers indicated that the two inbred strains differed on the genetic level. In order to further investigate whether these genetic differences have an impact on the disease phenotype of cystic fibrosis we characterised the phenotype of the two inbred strains.
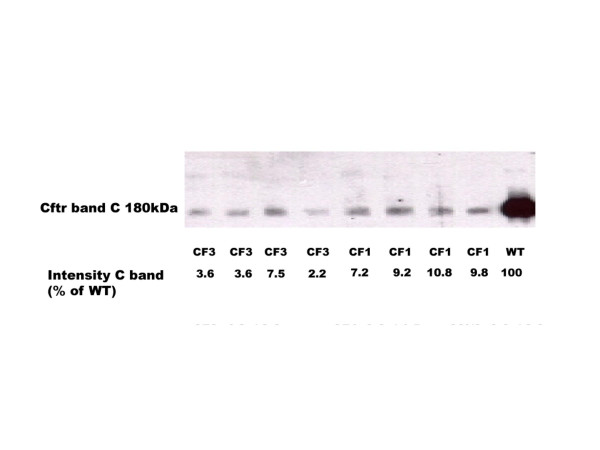
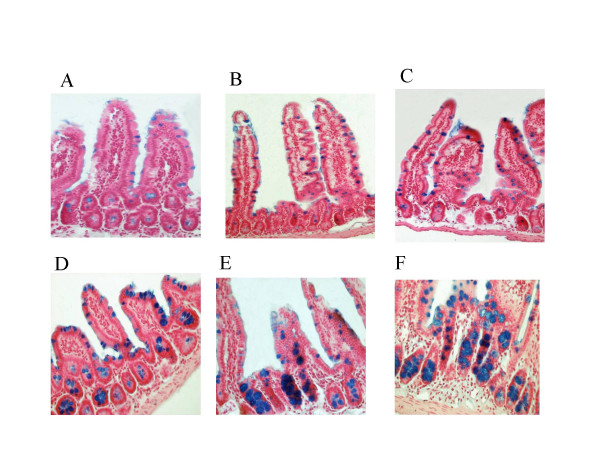
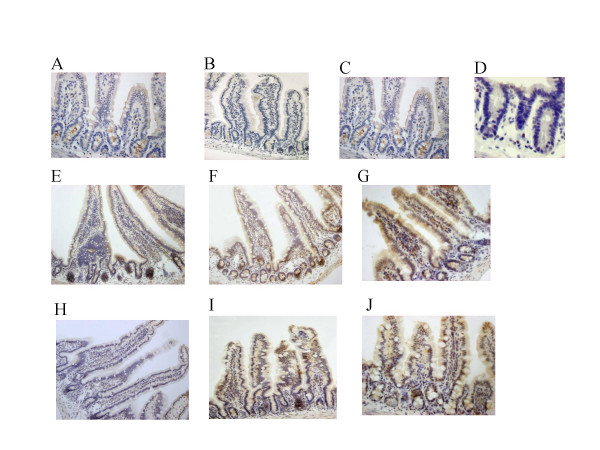
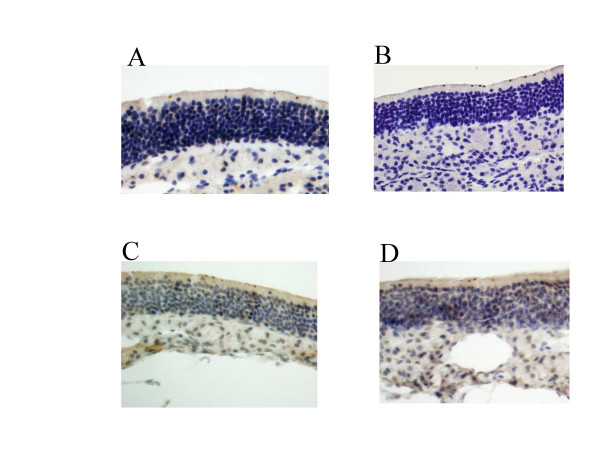
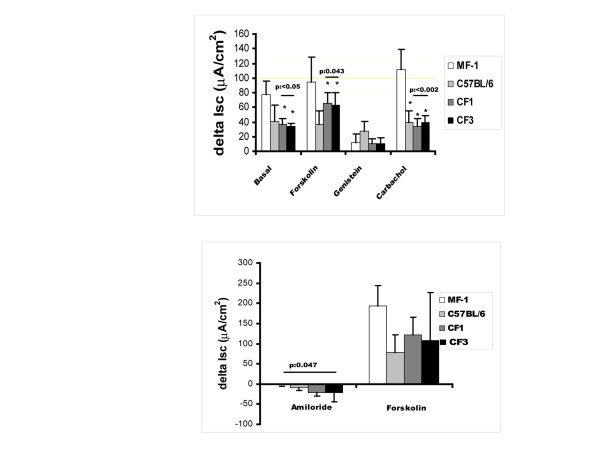
Results: Reduced amounts, compared to wild type control animals, of correctly spliced Cftr mRNA were detected in the nasal epithelia, lungs and the intestine of both inbred CftrTgH(neoim)Hgu strains, with higher residual amount observed for CF/1-CftrTgH(neoim)Hgu than CF/3-CftrTgH(neoim)Hgu for every investigated tissue. Accordingly the amounts of wild type Cftr protein in the intestine were 9% for CF/1-CftrTgH(neoim)Hgu and 4% for CF/3-CftrTgH(neoim)Hgu. Unlike the apparent strain and/or tissue specific regulation of Cftr mRNA splicing, short circuit current measurements in the respiratory and intestinal epithelium revealed that both strains have ameliorated the basic defect of cystic fibrosis with a presentation of a normal electrophysiology in both tissues.
Conclusion: Unlike the outbred CftrTgH(neoim)Hgu insertional mouse model, which displayed the electrophysiological defect in the gastrointestinal and respiratory tracts characteristic of cystic fibrosis, both inbred CftrTgH(neoim)Hgu strains have ameliorated the electrophysiological defect. On the basis of these findings both CF/1-CftrTgH(neoim)Hgu and CF/3-CftrTgH(neoim)Hgu offer an excellent model whereby determination of the minimal levels of protein required for the restoration of the basic defect of cystic fibrosis can be studied, along with the modulating factors which may affect this outcome.
Figures
Similar articles
-
Very mild disease phenotype of congenic CftrTgH(neoim)Hgu cystic fibrosis mice.BMC Genet. 2008 Apr 9;9:28. doi: 10.1186/1471-2156-9-28. BMC Genet. 2008. PMID: 18400105 Free PMC article.
-
Instability of the insertional mutation in CftrTgH(neoim)Hgu cystic fibrosis mouse model.BMC Genet. 2004 Apr 21;5:6. doi: 10.1186/1471-2156-5-6. BMC Genet. 2004. PMID: 15102331 Free PMC article.
-
Characterisation of electrogenic nutrient absorption in the Cftr TgH(neoim)Hgu mouse model.J Comp Physiol B. 2008 Aug;178(6):705-12. doi: 10.1007/s00360-008-0259-7. Epub 2008 Mar 28. J Comp Physiol B. 2008. PMID: 18369642
-
Mouse models of cystic fibrosis: phenotypic analysis and research applications.J Cyst Fibros. 2011 Jun;10 Suppl 2:S152-71. doi: 10.1016/S1569-1993(11)60020-9. J Cyst Fibros. 2011. PMID: 21658634 Review.
-
Genotype-phenotype correlation in cystic fibrosis: the role of modifier genes.Am J Med Genet. 2002 Jul 22;111(1):88-95. doi: 10.1002/ajmg.10461. Am J Med Genet. 2002. PMID: 12124743 Review.
Cited by
-
Multicenter intestinal current measurements in rectal biopsies from CF and non-CF subjects to monitor CFTR function.PLoS One. 2013 Sep 10;8(9):e73905. doi: 10.1371/journal.pone.0073905. eCollection 2013. PLoS One. 2013. PMID: 24040112 Free PMC article.
-
Characterisation of chloride currents across the proximal colon in CftrTgH(neoim)1Hgu congenic mice.J Comp Physiol B. 2007 Jan;177(1):61-73. doi: 10.1007/s00360-006-0109-4. Epub 2006 Jul 26. J Comp Physiol B. 2007. PMID: 16868751
-
Murine mCLCA6 is an integral apical membrane protein of non-goblet cell enterocytes and co-localizes with the cystic fibrosis transmembrane conductance regulator.J Histochem Cytochem. 2008 May;56(5):495-509. doi: 10.1369/jhc.2008.950592. Epub 2008 Feb 18. J Histochem Cytochem. 2008. PMID: 18285349 Free PMC article.
-
Very mild disease phenotype of congenic CftrTgH(neoim)Hgu cystic fibrosis mice.BMC Genet. 2008 Apr 9;9:28. doi: 10.1186/1471-2156-9-28. BMC Genet. 2008. PMID: 18400105 Free PMC article.
-
β1-Integrin Accumulates in Cystic Fibrosis Luminal Airway Epithelial Membranes and Decreases Sphingosine, Promoting Bacterial Infections.Cell Host Microbe. 2017 Jun 14;21(6):707-718.e8. doi: 10.1016/j.chom.2017.05.001. Epub 2017 May 25. Cell Host Microbe. 2017. PMID: 28552668 Free PMC article.
References
-
- Riordan J, Rommens J, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J, Drumm M, Iannuzzi M, Collins F, Tsui LC. Identification of the cystic fibrosis gene: cloning and characterisation of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Chiba-Falek O, Parad RB, Kerem E, Kerem B. Variable levels of normal RNA in different fetal organs carrying a cystic fibrosis transmembrane conductance regulator splicing mutation. Am J Respir Crit Care Med. 1999;159:1998–2002. - PubMed
-
- Ramalho AS, Beck S, Meyer M, Penque D, Cutting GR, Amaral MD. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am J Respir Cell Mol Biol. 2002;27:619–627. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
