

Identifying metabolic enzymes with multiple types of association evidence
- PMID: 16571130
- PMCID: PMC1450304
- DOI: 10.1186/1471-2105-7-177
Identifying metabolic enzymes with multiple types of association evidence
Abstract
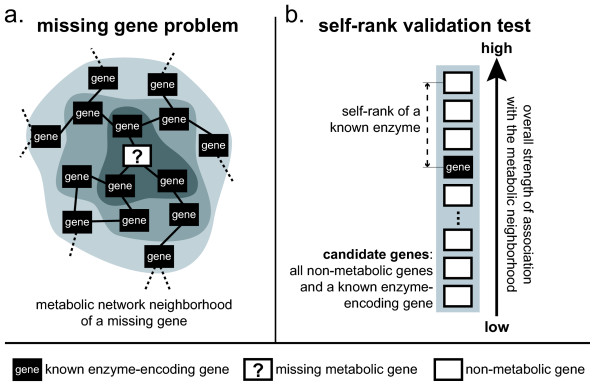
Background: Existing large-scale metabolic models of sequenced organisms commonly include enzymatic functions which can not be attributed to any gene in that organism. Existing computational strategies for identifying such missing genes rely primarily on sequence homology to known enzyme-encoding genes.
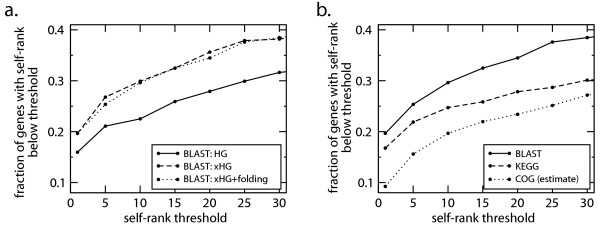
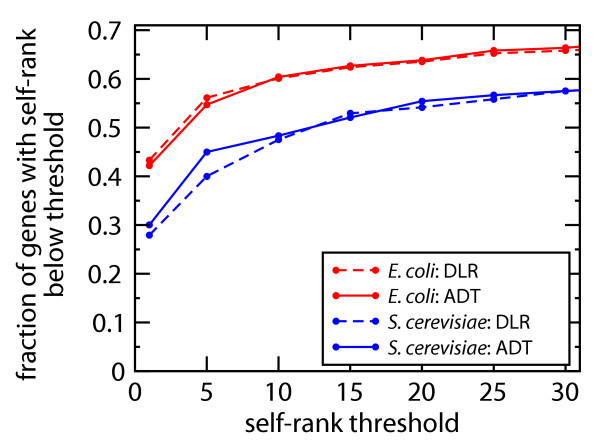
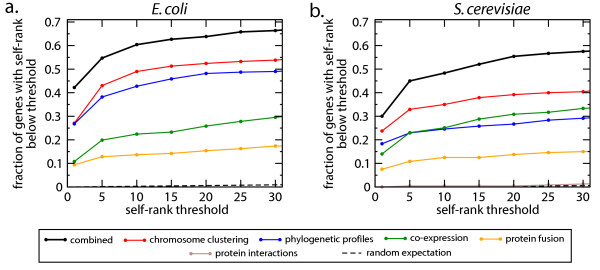
Results: We present a novel method for identifying genes encoding for a specific metabolic function based on a local structure of metabolic network and multiple types of functional association evidence, including clustering of genes on the chromosome, similarity of phylogenetic profiles, gene expression, protein fusion events and others. Using E. coli and S. cerevisiae metabolic networks, we illustrate predictive ability of each individual type of association evidence and show that significantly better predictions can be obtained based on the combination of all data. In this way our method is able to predict 60% of enzyme-encoding genes of E. coli metabolism within the top 10 (out of 3551) candidates for their enzymatic function, and as a top candidate within 43% of the cases.
Conclusion: We illustrate that a combination of genome context and other functional association evidence is effective in predicting genes encoding metabolic enzymes. Our approach does not rely on direct sequence homology to known enzyme-encoding genes, and can be used in conjunction with traditional homology-based metabolic reconstruction methods. The method can also be used to target orphan metabolic activities.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
