

Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema
- PMID: 16571143
- PMCID: PMC1501017
- DOI: 10.1186/1465-9921-7-53
Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema
Abstract
Chronic obstructive pulmonary disease (COPD) is characterised by chronic inflammation of the airways and progressive destruction of lung parenchyma, a process that in most cases is initiated by cigarette smoking. Several mechanisms are involved in the development of the disease: influx of inflammatory cells into the lung (leading to chronic inflammation of the airways), imbalance between proteolytic and anti-proteolytic activity (resulting in the destruction of healthy lung tissue) and oxidative stress. Recently, an increasing number of data suggest a fourth important mechanism involved in the development of COPD: apoptosis of structural cells in the lung might possibly be an important upstream event in the pathogenesis of COPD. There is an increase in apoptotic alveolar epithelial and endothelial cells in the lungs of COPD patients. Since this is not counterbalanced by an increase in proliferation of these structural cells, the net result is destruction of lung tissue and the development of emphysema. Data from animal models suggest a role for Vascular Endothelial Growth Factor (VEGF) in the induction of apoptosis of structural cells in the lung. Other mediators of apoptosis, such as caspase-3 and ceramide, could be interesting targets to prevent apoptosis and the development of emphysema. In this review, recent data on the role of apoptosis in COPD from both animal models as well as from studies on human subjects will be discussed. The aim is to provide an up to date summary on the increasing knowledge on the role of apoptosis in COPD and pulmonary emphysema.
Figures
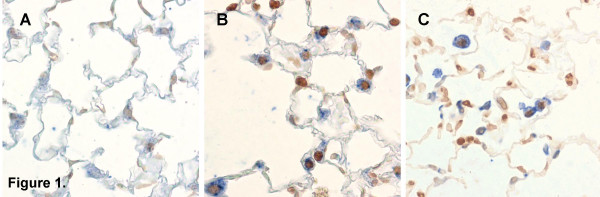
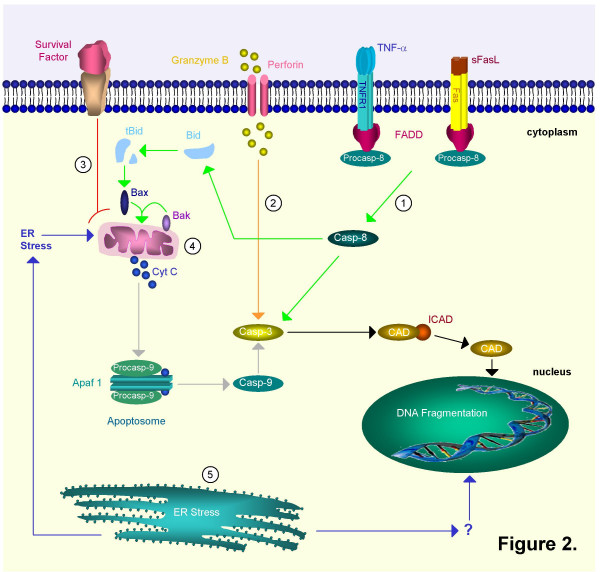
 Ligand-death-receptor pathway (green): death factors such as Fas ligand (FasL) and tumour necrosis factor (TNF) trigger apoptosis by binding on 'death receptors' such as Fas and Tumour Necrosis Factor Receptor 1 (TNFR1). FasL may be solubilized to sFasL by matrix metalloproteinases (MMP's). The death receptors recruit procaspase-8 by means of an adaptor protein, Fas associated death domain protein (FADD). After cleavage the mature caspase-8 then directly activates caspase-3 or cleaves Bid. Truncated Bid (tBid) interacts with Bax and Bak. A pore is formed in the outer mitochondrial membrane through which cytochrome c (Cyt C) is released. 2.
Ligand-death-receptor pathway (green): death factors such as Fas ligand (FasL) and tumour necrosis factor (TNF) trigger apoptosis by binding on 'death receptors' such as Fas and Tumour Necrosis Factor Receptor 1 (TNFR1). FasL may be solubilized to sFasL by matrix metalloproteinases (MMP's). The death receptors recruit procaspase-8 by means of an adaptor protein, Fas associated death domain protein (FADD). After cleavage the mature caspase-8 then directly activates caspase-3 or cleaves Bid. Truncated Bid (tBid) interacts with Bax and Bak. A pore is formed in the outer mitochondrial membrane through which cytochrome c (Cyt C) is released. 2.  Cytolytic effector cell pathway (orange): cytotoxic T cells can release granzyme B and perforin, a pore-forming protein. Granzyme B activates caspase-3 through cleavage. It can also cleave caspase-8. 3.
Cytolytic effector cell pathway (orange): cytotoxic T cells can release granzyme B and perforin, a pore-forming protein. Granzyme B activates caspase-3 through cleavage. It can also cleave caspase-8. 3.  Growth factor depletion pathway (red): deprivation of survival factors triggers Cyt C release through activation of Bax and Bak. Intrinsic pathway: 4.
Growth factor depletion pathway (red): deprivation of survival factors triggers Cyt C release through activation of Bax and Bak. Intrinsic pathway: 4.  Mitochondrial pathway (grey): mitochondria release cytochrome c (Cyt C) in response to stress. Together with apoptotic protease activating factor-1 (Apaf-1) and procaspase-9, Cyt C will form the apoptosome complex. This results in the proteolytic activation of the procaspase. Mature caspase-9 can then proteolytically activate caspase-3 and other executioner caspases. 5.
Mitochondrial pathway (grey): mitochondria release cytochrome c (Cyt C) in response to stress. Together with apoptotic protease activating factor-1 (Apaf-1) and procaspase-9, Cyt C will form the apoptosome complex. This results in the proteolytic activation of the procaspase. Mature caspase-9 can then proteolytically activate caspase-3 and other executioner caspases. 5.  Endoplasmatic reticulum pathway (blue): the ER can also induce apoptosis as a reaction to stress. It might do so by stimulating the mitochondrial pathway or by directly targeting the nucleus. In mice both caspase-7 and -12 are linked to this pathway. These different initiation pathways converge further downstream into activation of caspase-3. The effector caspase-3 cleaves ICAD (inhibitor of CAD) and releases it from CAD (caspase-activated DNAase). CAD translocates from the cytoplasm to the nucleus and can now act as active endonuclease and fragment DNA.
Endoplasmatic reticulum pathway (blue): the ER can also induce apoptosis as a reaction to stress. It might do so by stimulating the mitochondrial pathway or by directly targeting the nucleus. In mice both caspase-7 and -12 are linked to this pathway. These different initiation pathways converge further downstream into activation of caspase-3. The effector caspase-3 cleaves ICAD (inhibitor of CAD) and releases it from CAD (caspase-activated DNAase). CAD translocates from the cytoplasm to the nucleus and can now act as active endonuclease and fragment DNA.
References
-
- Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163:1256–1276. - PubMed
-
- Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22:672–688. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
