

Structure-guided recombination creates an artificial family of cytochromes P450
- PMID: 16594730
- PMCID: PMC1431580
- DOI: 10.1371/journal.pbio.0040112
Structure-guided recombination creates an artificial family of cytochromes P450
Abstract
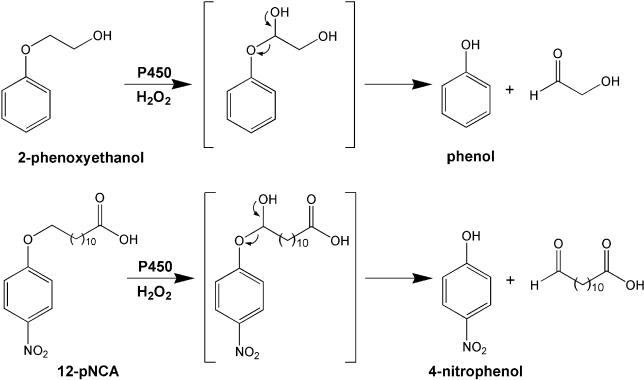
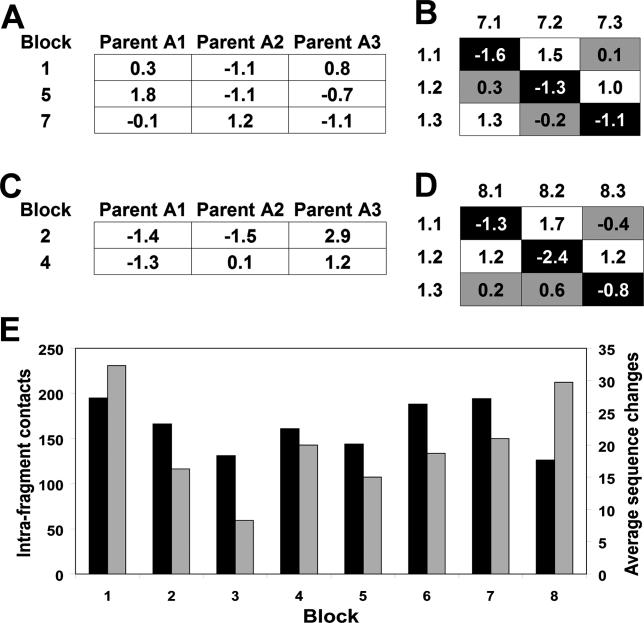
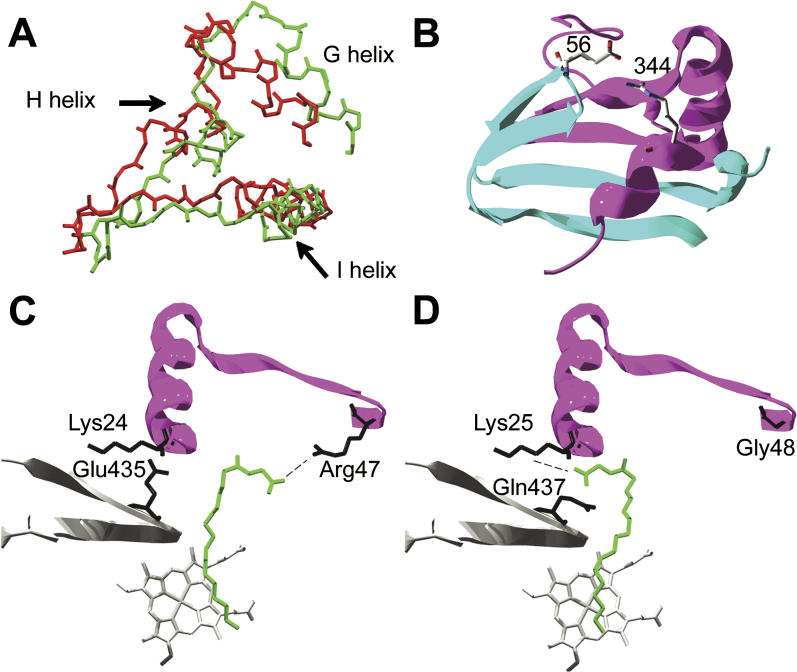
Creating artificial protein families affords new opportunities to explore the determinants of structure and biological function free from many of the constraints of natural selection. We have created an artificial family comprising 3,000 P450 heme proteins that correctly fold and incorporate a heme cofactor by recombining three cytochromes P450 at seven crossover locations chosen to minimize structural disruption. Members of this protein family differ from any known sequence at an average of 72 and by as many as 109 amino acids. Most (>73%) of the properly folded chimeric P450 heme proteins are catalytically active peroxygenases; some are more thermostable than the parent proteins. A multiple sequence alignment of 955 chimeras, including both folded and not, is a valuable resource for sequence-structure-function studies. Logistic regression analysis of the multiple sequence alignment identifies key structural contributions to cytochrome P450 heme incorporation and peroxygenase activity and suggests possible structural differences between parents CYP102A1 and CYP102A2.
Figures
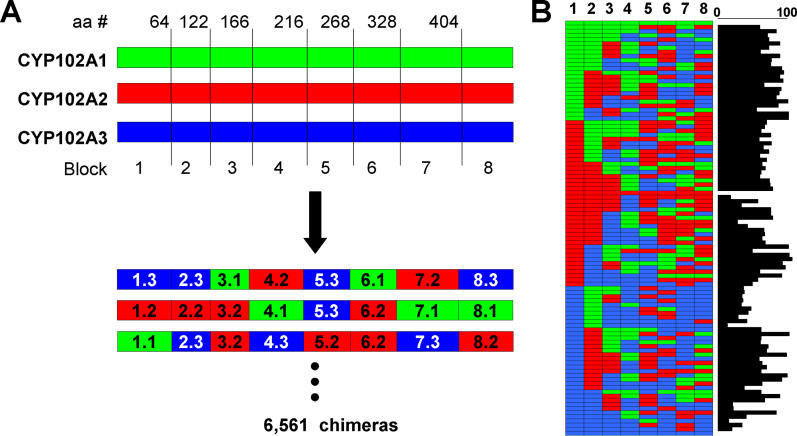
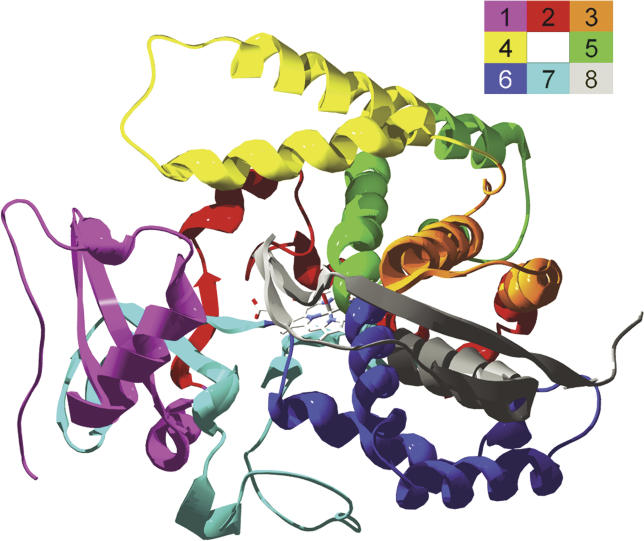
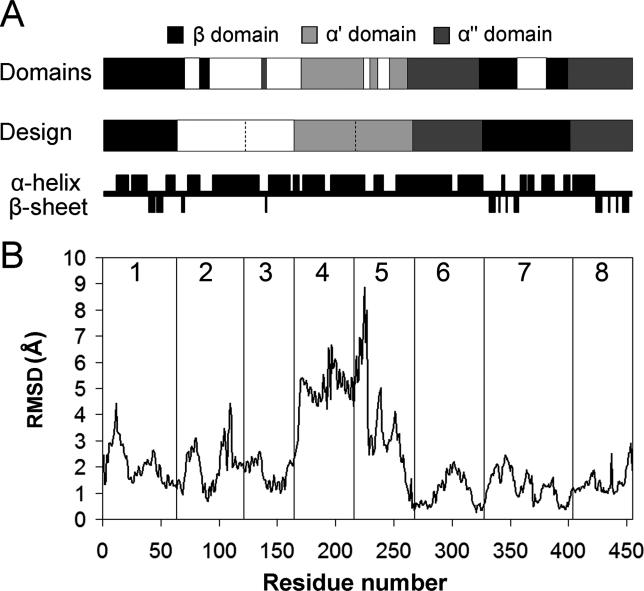
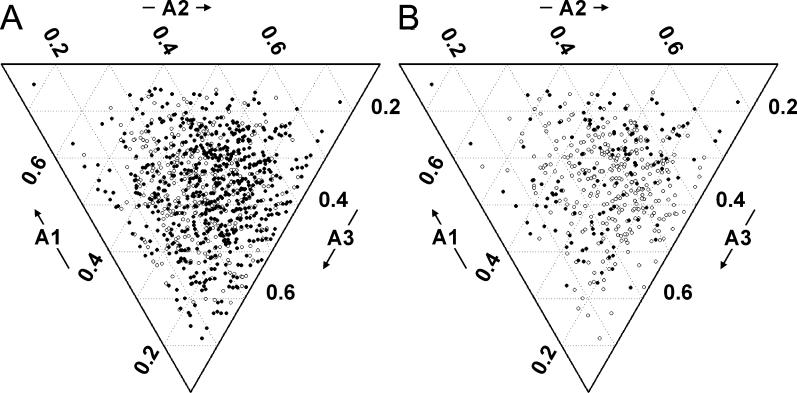
Comment in
-
A smart mutation scheme produces hundreds of functional proteins.PLoS Biol. 2006 May;4(5):e136. doi: 10.1371/journal.pbio.0040136. Epub 2006 Apr 11. PLoS Biol. 2006. PMID: 20076565 Free PMC article. No abstract available.
References
-
- Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. - PubMed
-
- Saraf MC, Moore GL, Maranas CD. Using multiple sequence correlation analysis to characterize functionally important protein regions. Protein Eng. 2003;16:397–406. - PubMed
-
- Moffet DA, Hecht MH. De novo proteins from combinatorial libraries. Chem Rev. 2001;101:3191–3203. - PubMed
-
- Arnold FH, Wintrode PL, Miyazaki K, Gershenson A. How enzymes adapt: Lessons from directed evolution. Trends Biochem Sci. 2001;26:100–106. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
