

Fabry disease: identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations
- PMID: 16595074
- PMCID: PMC3500179
- DOI: 10.1186/1479-7364-2-5-297
Fabry disease: identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations
Abstract
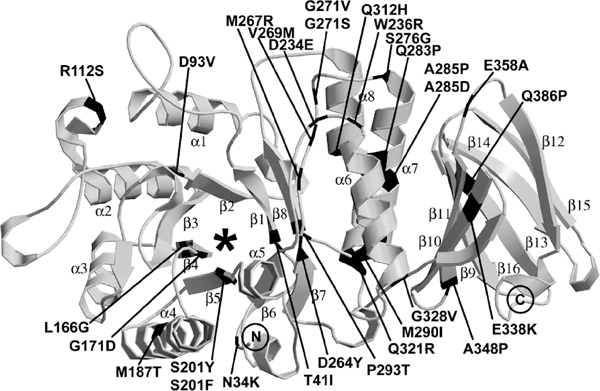
Fabry disease, an X-linked recessive inborn error of glycosphingolipid catabolism, results from the deficient activity of the lysosomal exoglycohydrolase, alpha-galactosidase A (EC 3.2.1.22; alpha-Gal A). The molecular lesions in the alpha-Gal A gene causing the classic phenotype of Fabry disease in 66 unrelated families were determined. In 49 families, 50 new mutations were identified, including: 29 missense mutations (N34K, T41I, D93V, R112S, L166G, G171D, M187T, S201Y, S201F, D234E, W236R, D264Y, M267R, V269M, G271S, G271V, S276G, Q283P, A285P, A285D, M290I, P293T, Q312H, Q321R, G328V, E338K, A348P, E358A, Q386P); nine nonsense mutations (C56X, E79X, K127X, Y151X, Y173X, L177X, W262X, Q306X, E338X); five splicing defects (IVS4-1G>A, IVS5-2A>G, IVS5+3A>G, IVS5+4A>G, IVS6-1G>C); four small deletions (18delA, 457delGAC, 567delG, 1096delACCAT); one small insertion (996insC); one 3.1 kilobase Alu-Alu deletion (which included exon 2); and one complex mutation (K374R, 1124delGAG). In 18 families, 17 previously reported mutations were identified, with R112C occurring in two families. In two classically affected families, affected males were identified with two mutations: one with two novel mutations, D264Y and V269M and the other with one novel (Q312H) and one previously reported (A143T) mutation. Transient expression of the individual mutations revealed that D264Y and Q312H were localised in the endoplasmic reticulum and had no detectable or markedly reduced activity, whereas V269M and A143T were localised in lysosomes and had approximately 10 per cent and approximately 35 per cent of expressed wild-type activity, respectively. Structural analyses based on the enzyme's three-dimensional structure predicted the effect of the 29 novel missense mutations on the mutant glycoprotein's structure. Of note, three novel mutations (approximately 10 per cent) were predicted not to significantly alter the glycoprotein's structure; however, they were disease causing. These studies further define the molecular heterogeneity of the alpha-Gal A mutations in classical Fabry disease, permit precise heterozygote detection and prenatal diagnosis, and provide insights into the structural alterations of the mutant enzymes that cause the classic phenotype.
Figures
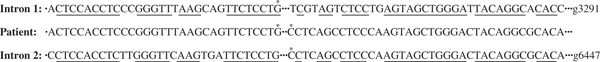
References
-
- Desnick RJ, Ioannou YA, Eng CM, In: The Metabolic and Molecular Bases of Inherited Disease. 8. Scriver CR, Beaudet AL, Sly WS, et al, editor. McGraw-Hill, New York, NY; 2001. α-Galactosidase A deficiency: Fabry disease; pp. 3733–3774.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
