

Multifactor dimensionality reduction: an analysis strategy for modelling and detecting gene-gene interactions in human genetics and pharmacogenomics studies
- PMID: 16595076
- PMCID: PMC3500181
- DOI: 10.1186/1479-7364-2-5-318
Multifactor dimensionality reduction: an analysis strategy for modelling and detecting gene-gene interactions in human genetics and pharmacogenomics studies
Abstract
The detection of gene-gene and gene-environment interactions associated with complex human disease or pharmacogenomic endpoints is a difficult challenge for human geneticists. Unlike rare, Mendelian diseases that are associated with a single gene, most common diseases are caused by the non-linear interaction of numerous genetic and environmental variables. The dimensionality involved in the evaluation of combinations of many such variables quickly diminishes the usefulness of traditional, parametric statistical methods. Multifactor dimensionality reduction (MDR) is a novel and powerful statistical tool for detecting and modelling epistasis. MDR is a non-parametric and model-free approach that has been shown to have reasonable power to detect epistasis in both theoretical and empirical studies. MDR has detected interactions in diseases such as sporadic breast cancer, multiple sclerosis and essential hypertension. As this method is more frequently applied, and was gained acceptance in the study of human disease and pharmacogenomics, it is becoming increasingly important that the implementation of the MDR approach is properly understood. As with all statistical methods, MDR is only powerful and useful when implemented correctly. Concerns regarding dataset structure, configuration parameters and the proper execution of permutation testing in reference to a particular dataset and configuration are essential to the method's effectiveness. The detection, characterisation and interpretation of gene-gene and gene-environment interactions are expected to improve the diagnosis, prevention and treatment of common human diseases. MDR can be a powerful tool in reaching these goals when used appropriately.
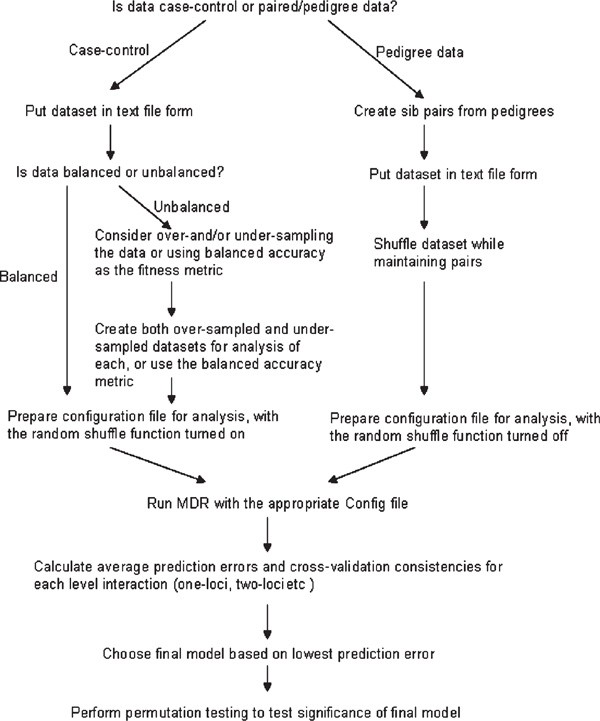
Figures



References
-
- Sing CF, Stengard JH, Kardia SL. Dynamic relationships between the genome and exposures to environments as causes of common human diseases. World Rev Nutr Diet. 2004;93:77–91. - PubMed
-
- Bellman R. Adaptive Control Processes. Princeton University Press, Princeton, WJ; 1961.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
