

Algorithms for incorporating prior topological information in HMMs: application to transmembrane proteins
- PMID: 16597327
- PMCID: PMC1523218
- DOI: 10.1186/1471-2105-7-189
Algorithms for incorporating prior topological information in HMMs: application to transmembrane proteins
Abstract
Background: Hidden Markov Models (HMMs) have been extensively used in computational molecular biology, for modelling protein and nucleic acid sequences. In many applications, such as transmembrane protein topology prediction, the incorporation of limited amount of information regarding the topology, arising from biochemical experiments, has been proved a very useful strategy that increased remarkably the performance of even the top-scoring methods. However, no clear and formal explanation of the algorithms that retains the probabilistic interpretation of the models has been presented so far in the literature.
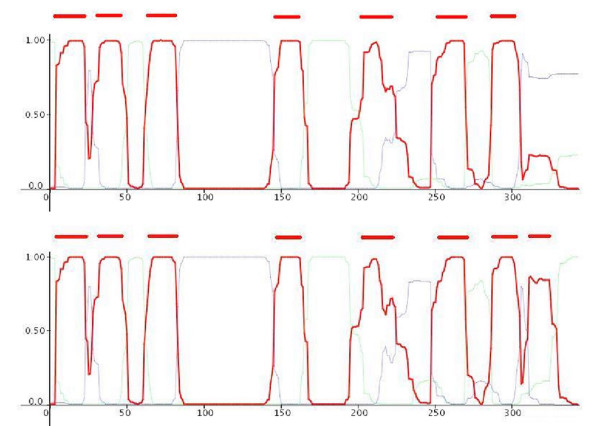
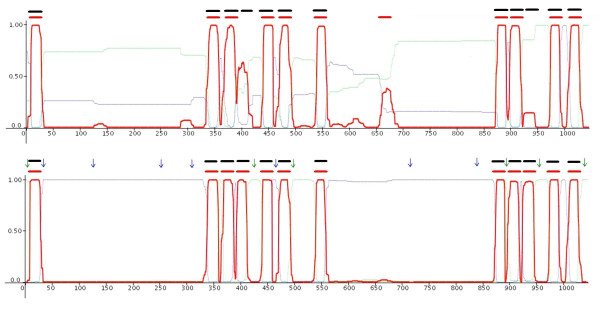
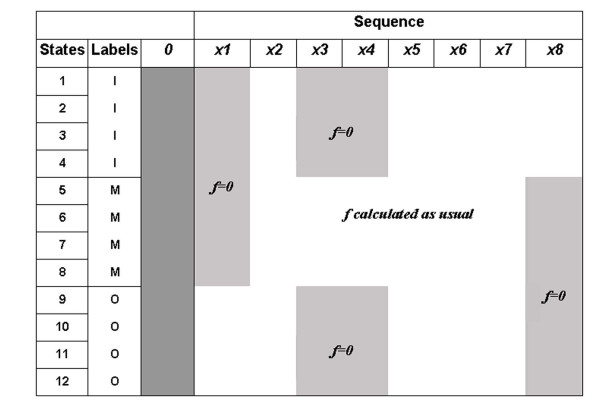
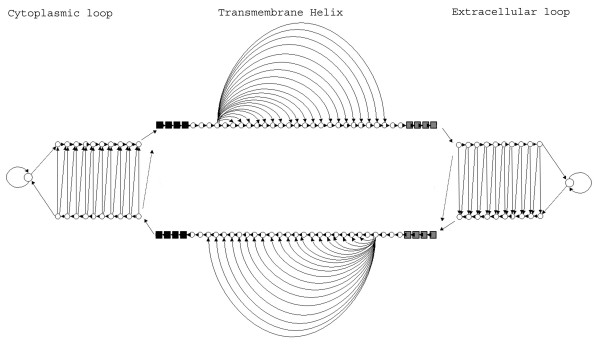
Results: We present here, a simple method that allows incorporation of prior topological information concerning the sequences at hand, while at the same time the HMMs retain their full probabilistic interpretation in terms of conditional probabilities. We present modifications to the standard Forward and Backward algorithms of HMMs and we also show explicitly, how reliable predictions may arise by these modifications, using all the algorithms currently available for decoding HMMs. A similar procedure may be used in the training procedure, aiming at optimizing the labels of the HMM's classes, especially in cases such as transmembrane proteins where the labels of the membrane-spanning segments are inherently misplaced. We present an application of this approach developing a method to predict the transmembrane regions of alpha-helical membrane proteins, trained on crystallographically solved data. We show that this method compares well against already established algorithms presented in the literature, and it is extremely useful in practical applications.
Conclusion: The algorithms presented here, are easily implemented in any kind of a Hidden Markov Model, whereas the prediction method (HMM-TM) is freely available for academic users at http://bioinformatics.biol.uoa.gr/HMM-TM, offering the most advanced decoding options currently available.
Figures
References
-
- Rabiner LR. A tutorial on hidden Markov models and selected applications in speech recognition. Proc IEEE. 1989;77: 257–286.
-
- Durbin R, Eddy SR, Krogh A, Mithison G. Biological sequence analysis, probabilistic models of proteins and nucleic acids. Cambridge University Press; 1998.
-
- Eddy SR. Multiple alignment using hidden Markov models. Proc Int Conf Intell Syst Mol Biol. 1995;3:114–120. - PubMed
-
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
