

Inclusion body formation and neurodegeneration are parkin independent in a mouse model of alpha-synucleinopathy
- PMID: 16597723
- PMCID: PMC6674122
- DOI: 10.1523/JNEUROSCI.0414-06.2006
Inclusion body formation and neurodegeneration are parkin independent in a mouse model of alpha-synucleinopathy
Abstract
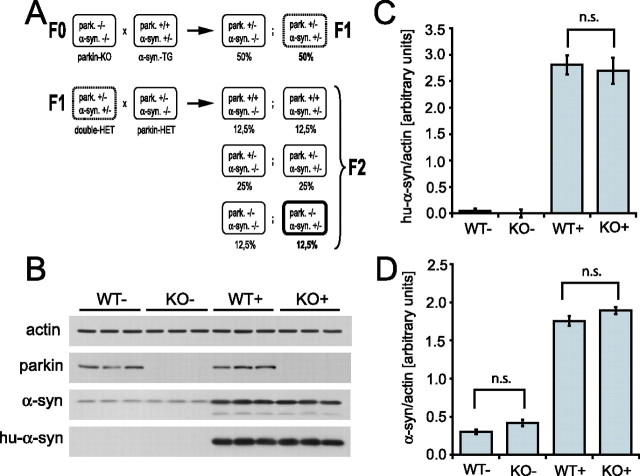
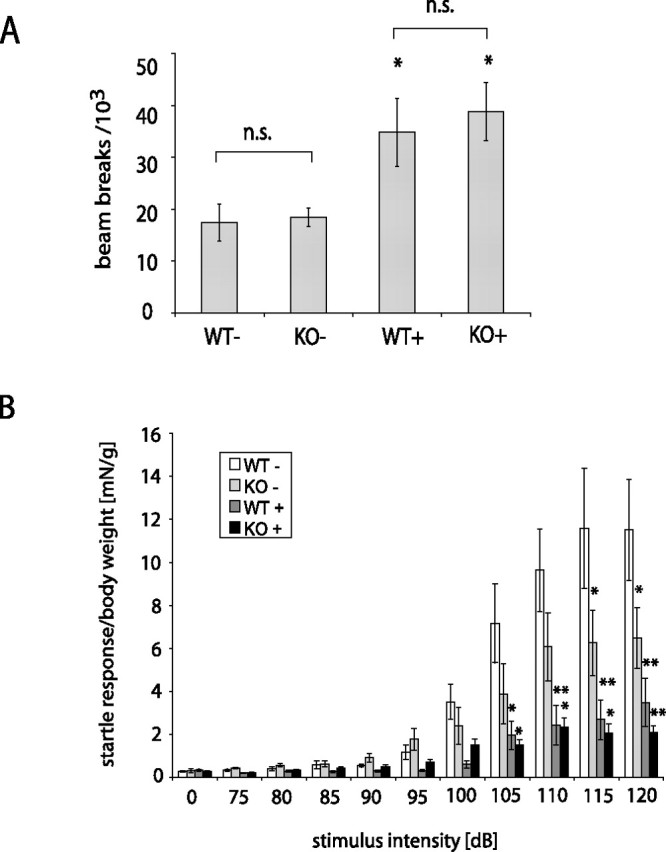
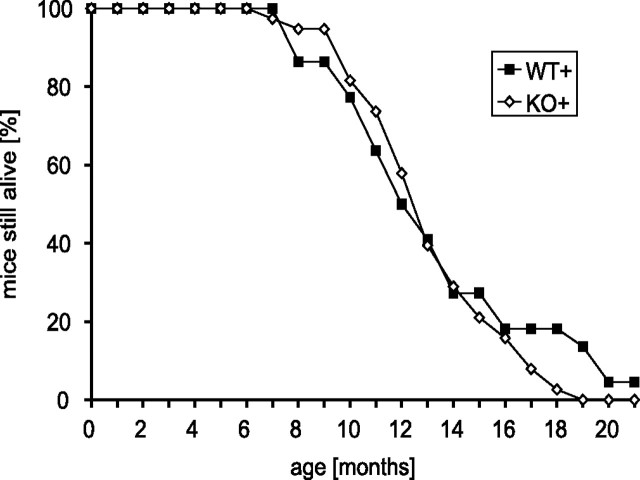
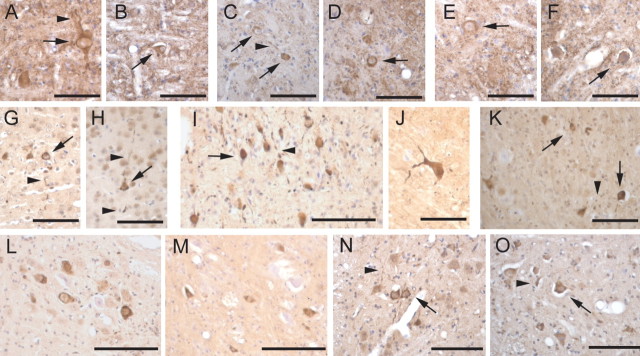
Mutations in the genes coding for alpha-synuclein and parkin cause autosomal-dominant and autosomal-recessive forms of Parkinson's disease (PD), respectively. Alpha-synuclein is a major component of Lewy bodies, the proteinaceous cytoplasmic inclusions that are the pathological hallmark of idiopathic PD. Lewy bodies appear to be absent in cases of familial PD associated with mutated forms of parkin. Parkin is an ubiquitin E3 ligase, and it may be involved in the processing and/or degradation of alpha-synuclein, as well as in the formation of Lewy bodies. Here we report the behavioral, biochemical, and histochemical characterization of double-mutant mice overexpressing mutant human A53T alpha-synuclein on a parkin null background. We find that the absence of parkin does not have an impact on the onset and progression of the lethal phenotype induced by overexpression of human A53T alpha-synuclein. Furthermore, all major behavioral, biochemical, and morphological characteristics of A53T alpha-synuclein-overexpressing mice are not altered in parkin null alpha-synuclein-overexpressing double-mutant mice. Our results demonstrate that mutant alpha-synuclein induces neurodegeneration independent of parkin-mediated ubiquitin E3 ligase activity in nondopaminergic systems and suggest that PD caused by alpha-synuclein and parkin mutations may occur via independent mechanisms.
Figures
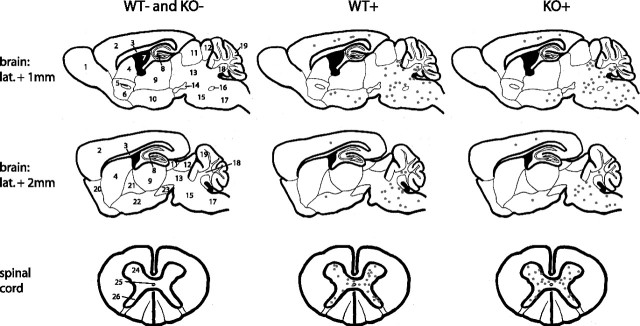
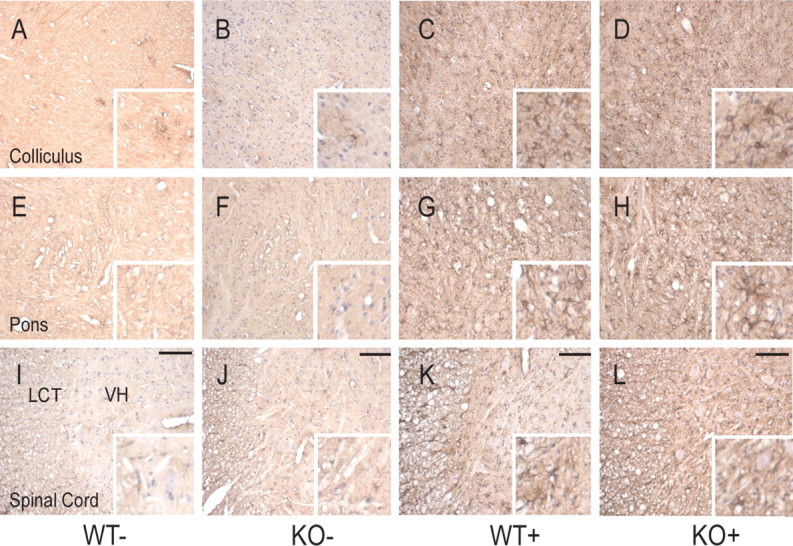
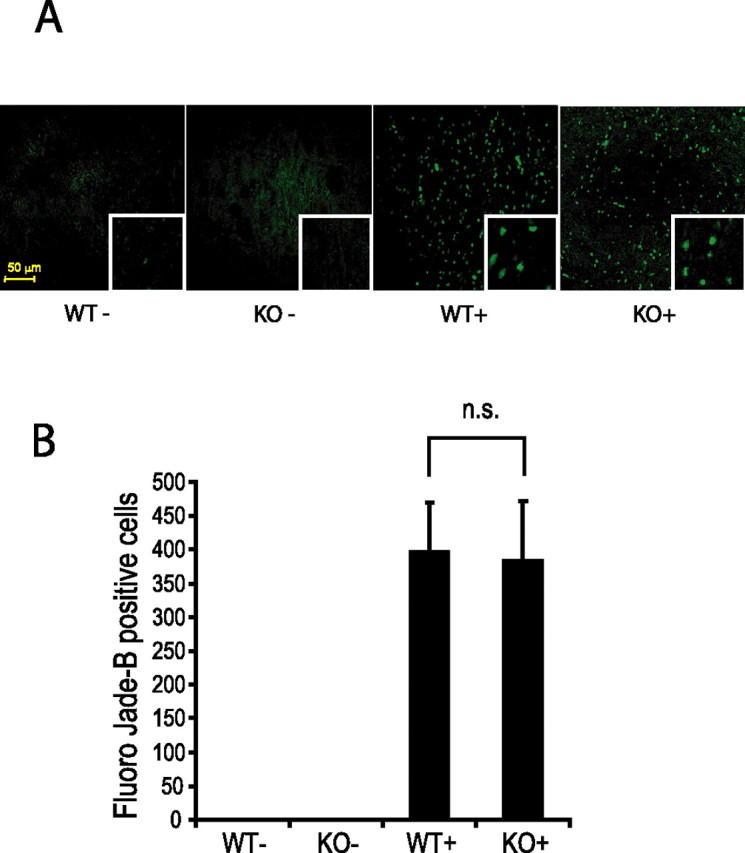
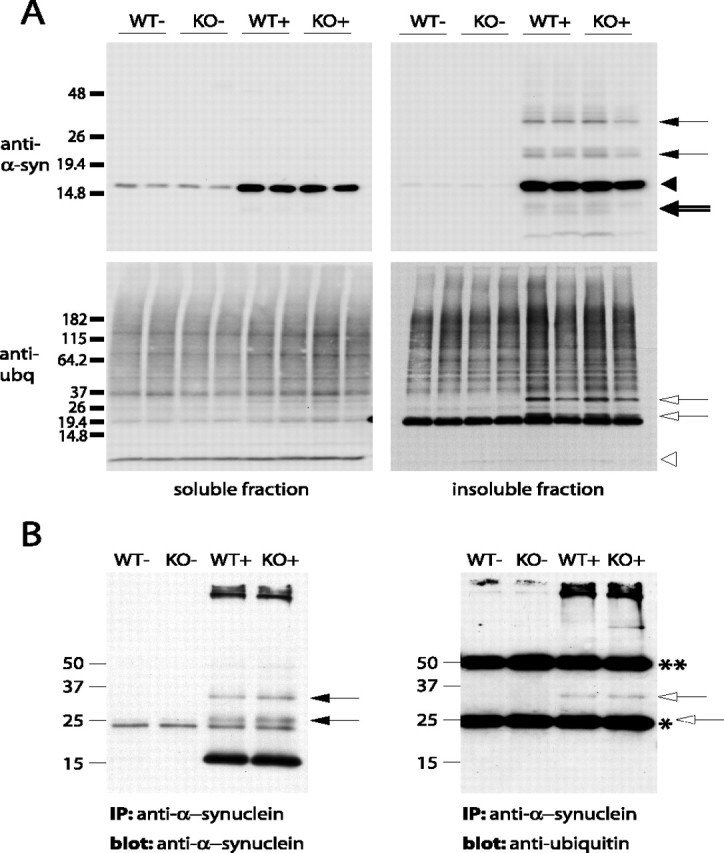
References
-
- Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM (1999). Degradation of alpha-synuclein by proteasome. J Biol Chem 274:33855–33858. - PubMed
-
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. - PubMed
-
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM (2001). Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med 7:1144–1150. - PubMed
-
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM (2004). S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science 304:1328–1331. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases