

A KSR/CNK complex mediated by HYP, a novel SAM domain-containing protein, regulates RAS-dependent RAF activation in Drosophila
- PMID: 16600912
- PMCID: PMC1472284
- DOI: 10.1101/gad.1390406
A KSR/CNK complex mediated by HYP, a novel SAM domain-containing protein, regulates RAS-dependent RAF activation in Drosophila
Abstract
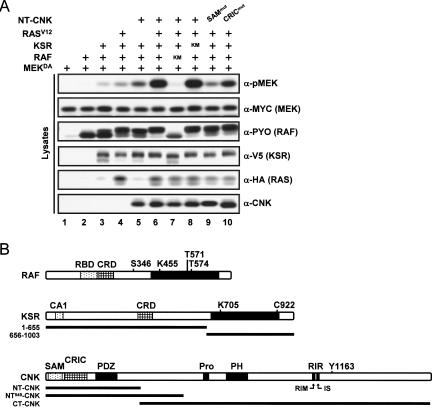
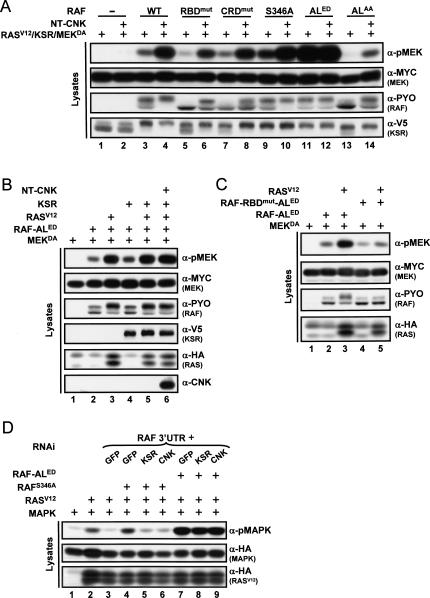
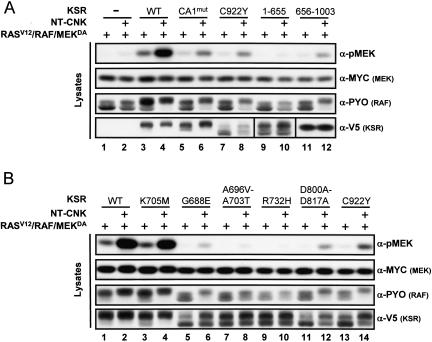
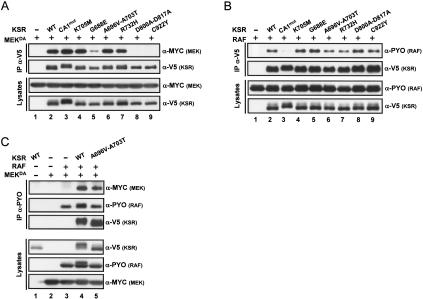
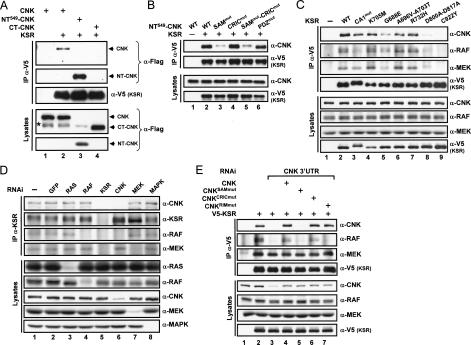
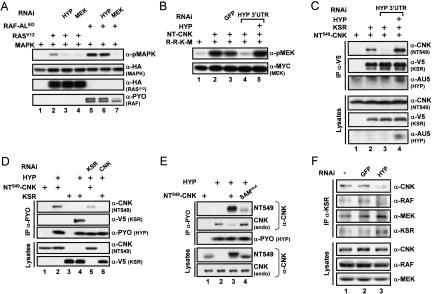
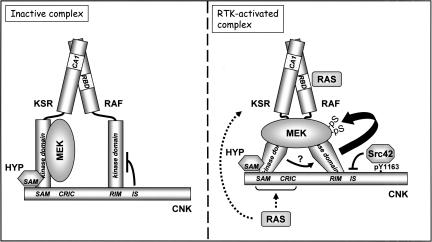
RAF is a critical effector of the small GTPase RAS in normal and malignant cells. Despite intense scrutiny, the mechanism regulating RAF activation remains partially understood. Here, we show that the scaffold KSR (kinase suppressor of RAS), a RAF homolog known to assemble RAF/MEK/ERK complexes, induces RAF activation in Drosophila by a mechanism mediated by its kinase-like domain, but which is independent of its scaffolding property or putative kinase activity. Interestingly, we found that KSR is recruited to RAF prior to signal activation by the RAF-binding protein CNK (connector enhancer of KSR) in association with a novel SAM (sterile alpha motif) domain-containing protein, named Hyphen (HYP). Moreover, our data suggest that the interaction of KSR to CNK/HYP stimulates the RAS-dependent RAF-activating property of KSR. Together, these findings identify a novel protein complex that controls RAF activation and suggest that KSR does not only act as a scaffold for the MAPK (mitogen-activated protein kinase) module, but may also function as a RAF activator. By analogy to catalytically impaired, but conformationally active B-RAF oncogenic mutants, we discuss the possibility that KSR represents a natural allosteric inducer of RAF catalytic function.
Figures
References
-
- Anselmo A.N., Bumeister R., Thomas J.M., White M.A. Critical contribution of linker proteins to Raf kinase activation. J. Biol. Chem. 2002;277:5940–5943. - PubMed
-
- Baker D.A., Mille-Baker B., Wainwright S.M., Ish-Horowicz D., Dibb N.J. Mae mediates MAP kinase phosphorylation of Ets transcription factors in Drosophila. Nature. 2001;411:330–334. - PubMed
-
- Bumeister R., Rosse C., Anselmo A., Camonis J., White M.A. CNK2 couples NGF signal propagation to multiple regulatory cascades driving cell differentiation. Curr. Biol. 2004;14:439–445. - PubMed
-
- Chong H., Vikis H.G., Guan K.L. Mechanisms of regulating the Raf kinase family. Cell. Signal. 2003;15:463–469. - PubMed
-
- Davies H., Bignell G.R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M.J., Bottomley W., et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous