

Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients
- PMID: 16600992
- PMCID: PMC2563157
- DOI: 10.1093/hmg/ddl084
Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients
Abstract
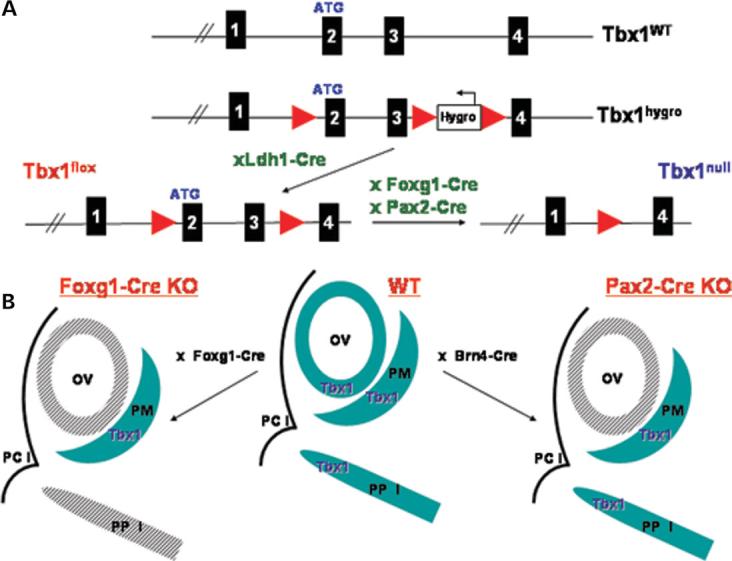
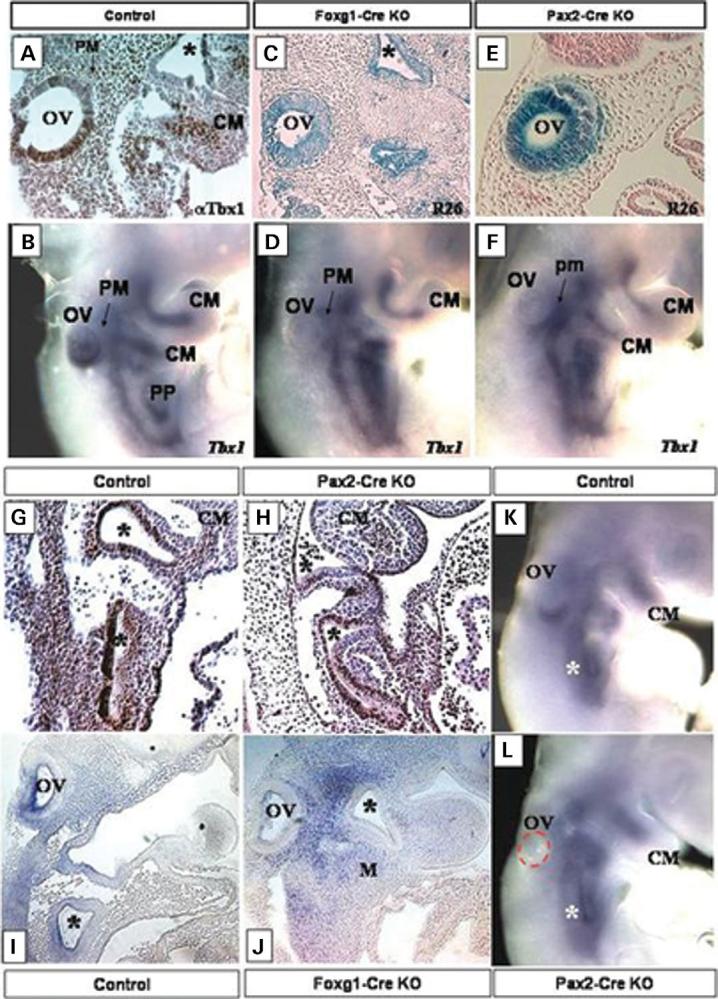
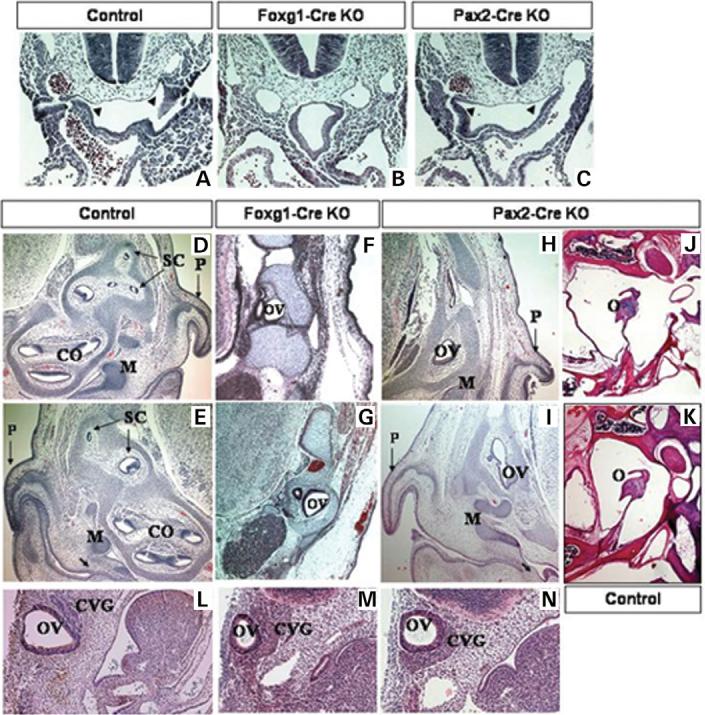
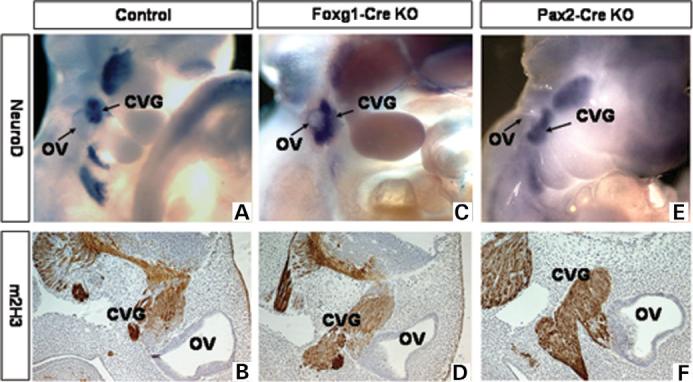
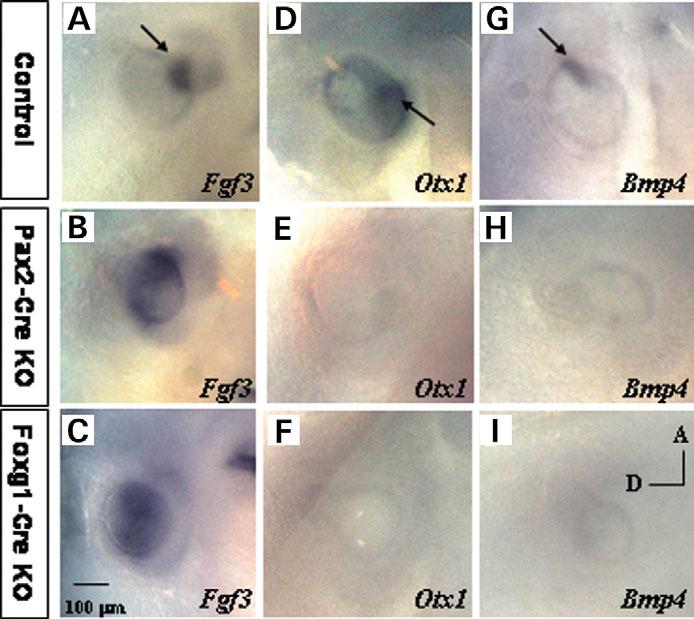
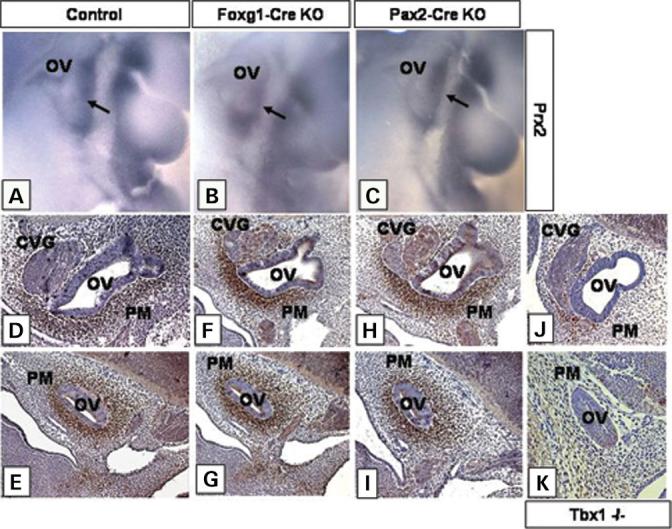
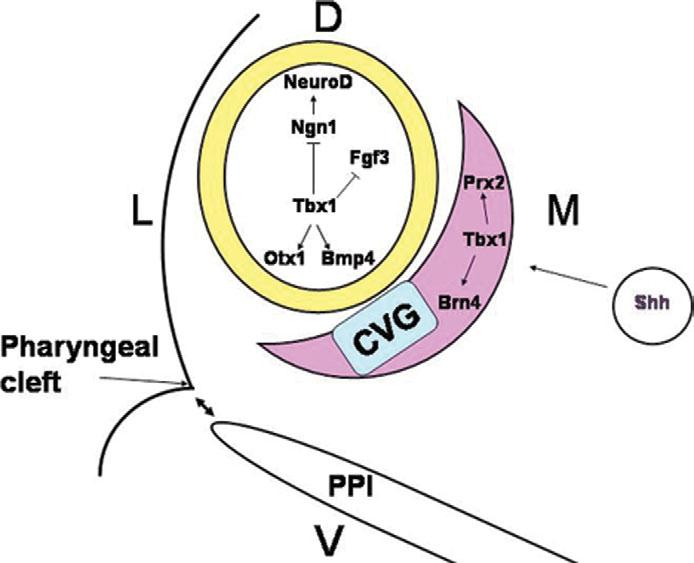
Most 22q11.2 deletion syndrome (22q11DS) patients have middle and outer ear anomalies, whereas some have inner ear malformations. Tbx1, a gene hemizygously deleted in 22q11DS patients and required for ear development, is expressed in multiple tissues during embryogenesis. To determine the role of Tbx1 in the first pharyngeal pouch (PPI) in forming outer and middle ears, we tissue-specifically inactivated the gene using Foxg1-Cre. In the conditional mutants, PPI failed to outgrow, preventing the middle ear bone condensations from forming. Tbx1 was also inactivated in the otic vesicle (OV), resulting in the failure of inner ear sensory organ formation, and in duplication of the cochleovestibular ganglion (CVG). Consistent with the anatomical defects, the sensory genes, Otx1 and Bmp4 were downregulated, whereas the CVG genes, Fgf3 and NeuroD, were upregulated. To delineate Tbx1 cell-autonomous roles, a more selective ablation, exclusively in the OV, was performed using Pax2-Cre. In contrast to the Foxg1-Cre mutants, Pax2-Cre conditional mutant mice survived to adulthood and had normal outer and middle ears but had the same inner ear defects as the Tbx1 null mice, with the same gene expression changes. These results demonstrate that Tbx1 has non-cell autonomous roles in PPI in the formation of outer and middle ears and cell-autonomous roles in the OV. Periotic mesenchymal markers, Prx2 and Brn4 were normal in both conditional mutants, whereas they were diminished in Tbx1-/- embryos. Thus, Tbx1 in the surrounding mesenchyme in both sets of conditional mutants cannot suppress the defects in the OV that occur in the null mutants.
Figures
References
-
- DiGeorge A. A new concept of the cellular basis of immunity. J. Pediatr. 1965;67:907.
-
- Emanuel BS, McDonald-McGinn D, Saitta SC, Zackai EH. The 22q11.2 deletion syndrome. Adv. Pediatr. 2001;48:39–73. - PubMed
-
- McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Solot C, Wang P, Jacobs I, et al. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Genet. Couns. 1999;10:11–24. - PubMed
-
- Greenberg F. What defines DiGeorge anomaly? J. Pediatr. 1989;115:412–413. - PubMed
-
- Schuknecht HF. Mondini dysplasia: a clinical and pathological study. Ann. Otol. Rhinol. Laryngol. Suppl. 1980;89:1–23. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
