

Tissue expression of PD-L1 mediates peripheral T cell tolerance
- PMID: 16606670
- PMCID: PMC2118286
- DOI: 10.1084/jem.20051776
Tissue expression of PD-L1 mediates peripheral T cell tolerance
Abstract
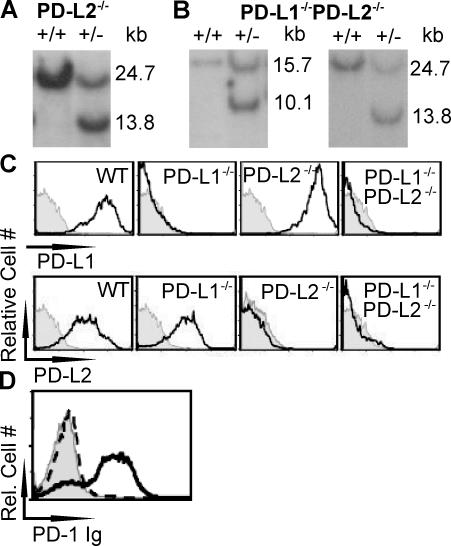
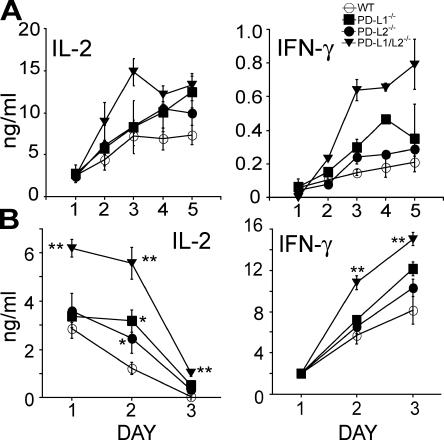
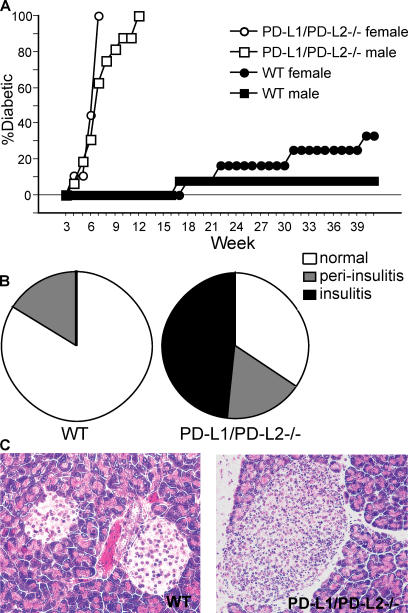
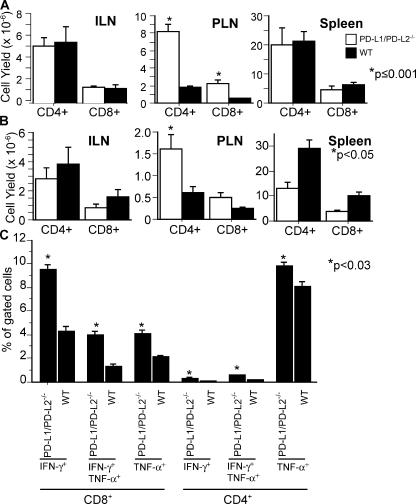
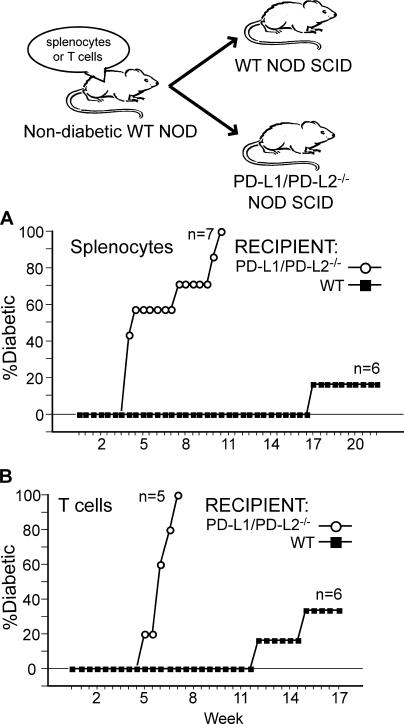
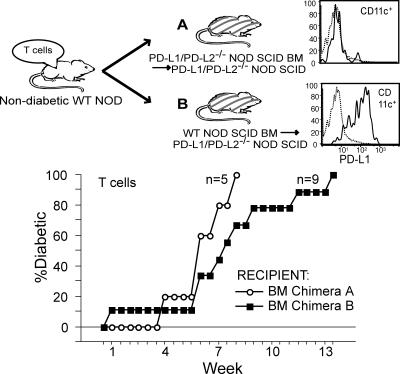
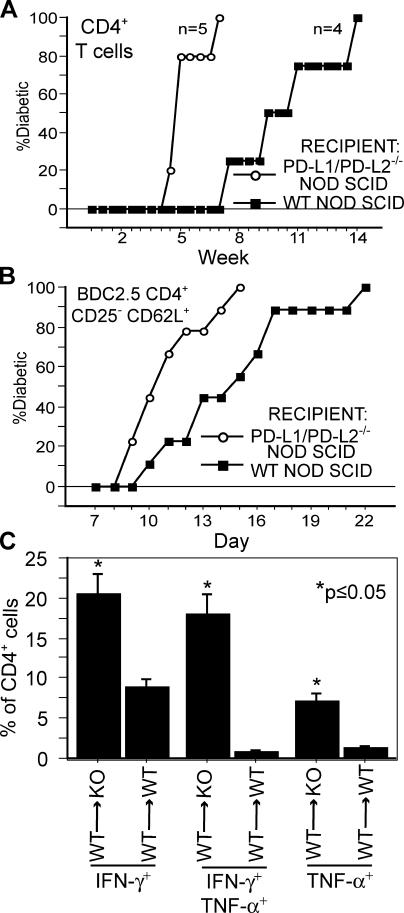
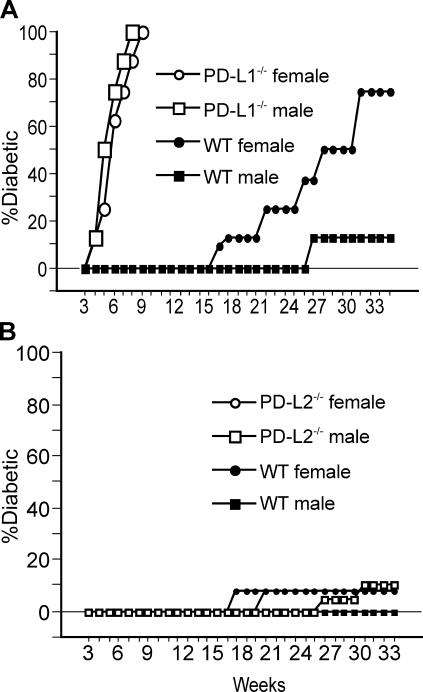
Programmed death 1 (PD-1), an inhibitory receptor expressed on activated lymphocytes, regulates tolerance and autoimmunity. PD-1 has two ligands: PD-1 ligand 1 (PD-L1), which is expressed broadly on hematopoietic and parenchymal cells, including pancreatic islet cells; and PD-L2, which is restricted to macrophages and dendritic cells. To investigate whether PD-L1 and PD-L2 have synergistic or unique roles in regulating T cell activation and tolerance, we generated mice lacking PD-L1 and PD-L2 (PD-L1/PD-L2(-/-) mice) and compared them to mice lacking either PD-L. PD-L1 and PD-L2 have overlapping functions in inhibiting interleukin-2 and interferon-gamma production during T cell activation. However, PD-L1 has a unique and critical role in controlling self-reactive T cells in the pancreas. Our studies with bone marrow chimeras demonstrate that PD-L1/PD-L2 expression only on antigen-presenting cells is insufficient to prevent the early onset diabetes that develops in PD-L1/PD-L2(-/-) non-obese diabetic mice. PD-L1 expression in islets protects against immunopathology after transplantation of syngeneic islets into diabetic recipients. PD-L1 inhibits pathogenic self-reactive CD4+ T cell-mediated tissue destruction and effector cytokine production. These data provide evidence that PD-L1 expression on parenchymal cells rather than hematopoietic cells protects against autoimmune diabetes and point to a novel role for PD-1-PD-L1 interactions in mediating tissue tolerance.
Figures
Comment in
-
New battlefields for costimulation.J Exp Med. 2006 Apr 17;203(4):817-20. doi: 10.1084/jem.20060219. Epub 2006 Apr 10. J Exp Med. 2006. PMID: 16606678 Free PMC article. Review.
References
-
- Anderson, M.S., and J.A. Bluestone. 2005. The NOD mouse: a model of immune dysregulation. Annu. Rev. Immunol. 23:447–485. - PubMed
-
- Keir, M.E., and A.H. Sharpe. 2005. The B7/CD28 costimulatory family in autoimmunity. Immunol. Rev. 204:128–143. - PubMed
-
- Chen, L. 2004. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 4:336–347. - PubMed
-
- Greenwald, R.J., Y.E. Latchman, and A.H. Sharpe. 2002. Negative co-receptors on lymphocytes. Curr. Opin. Immunol. 14:391–396. - PubMed
-
- Chambers, C.A., M.S. Kuhns, J.G. Egen, and J.P. Allison. 2001. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 19:565–594. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
