

Probing genomic diversity and evolution of Escherichia coli O157 by single nucleotide polymorphisms
- PMID: 16606700
- PMCID: PMC1473186
- DOI: 10.1101/gr.4759706
Probing genomic diversity and evolution of Escherichia coli O157 by single nucleotide polymorphisms
Abstract
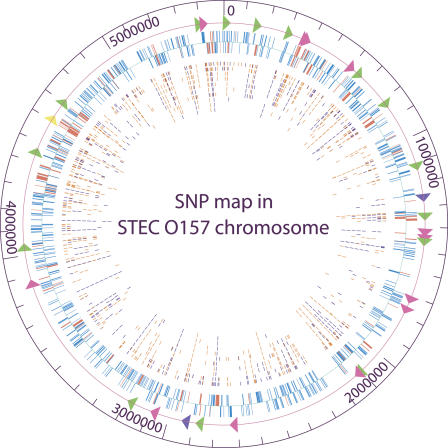
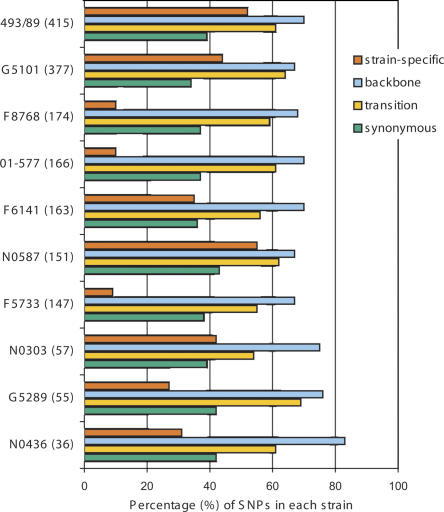
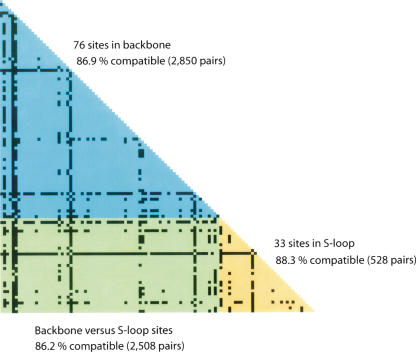
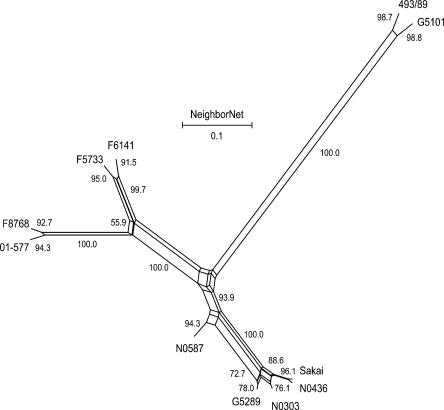
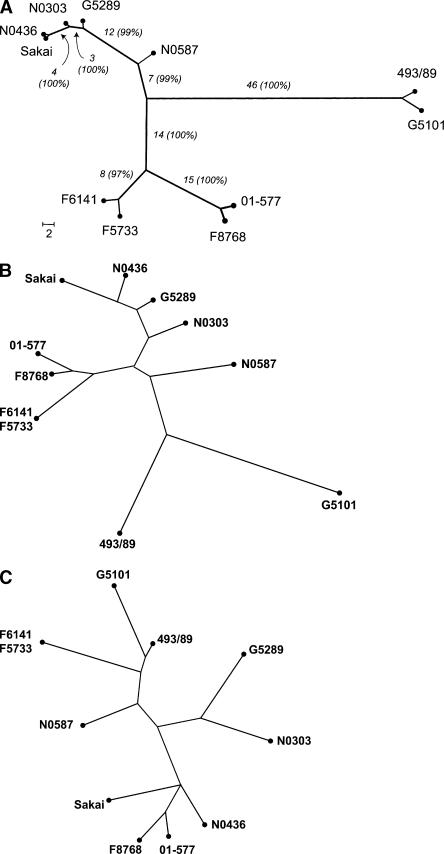
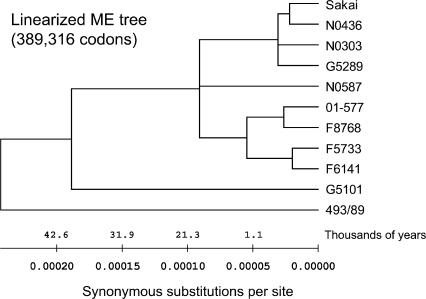
Infections by Shiga toxin-producing Escherichia coli O157:H7 (STEC O157) are the predominant cause of bloody diarrhea and hemolytic uremic syndrome in the United States. In silico comparison of the two complete STEC O157 genomes (Sakai and EDL933) revealed a strikingly high level of sequence identity in orthologous protein-coding genes, limiting the use of nucleotide sequences to study the evolution and epidemiology of this bacterial pathogen. To systematically examine single nucleotide polymorphisms (SNPs) at a genome scale, we designed comparative genome sequencing microarrays and analyzed 1199 chromosomal genes (a total of 1,167,948 bp) and 92,721 bp of the large virulence plasmid (pO157) of eleven outbreak-associated STEC O157 strains. We discovered 906 SNPs in 523 chromosomal genes and observed a high level of DNA polymorphisms among the pO157 plasmids. Based on a uniform rate of synonymous substitution for Escherichia coli and Salmonella enterica (4.7x10(-9) per site per year), we estimate that the most recent common ancestor of the contemporary beta-glucuronidase-negative, non-sorbitolfermenting STEC O157 strains existed ca. 40 thousand years ago. The phylogeny of the STEC O157 strains based on the informative synonymous SNPs was compared to the maximum parsimony trees inferred from pulsed-field gel electrophoresis and multilocus variable numbers of tandem repeats analysis. The topological discrepancies indicate that, in contrast to the synonymous mutations, parts of STEC O157 genomes have evolved through different mechanisms with highly variable divergence rates. The SNP loci reported here will provide useful genetic markers for developing high-throughput methods for fine-resolution genotyping of STEC O157. Functional characterization of nucleotide polymorphisms should shed new insights on the evolution, epidemiology, and pathogenesis of STEC O157 and related pathogens.
Figures
References
-
- Albert T.J., Norton J., Ott M., Richmond T., Nuwaysir K., Nuwaysir E.F., Stengele K.P., Green R.D., Norton J., Ott M., Richmond T., Nuwaysir K., Nuwaysir E.F., Stengele K.P., Green R.D., Ott M., Richmond T., Nuwaysir K., Nuwaysir E.F., Stengele K.P., Green R.D., Richmond T., Nuwaysir K., Nuwaysir E.F., Stengele K.P., Green R.D., Nuwaysir K., Nuwaysir E.F., Stengele K.P., Green R.D., Nuwaysir E.F., Stengele K.P., Green R.D., Stengele K.P., Green R.D., Green R.D. Light-directed 5′→3′ synthesis of complex oligonucleotide microarrays. Nucleic Acids Res. 2003;31:35–44. - PMC - PubMed
-
- Albert T.J., Dailidiene D., Dailide G., Norton J.E., Kalia A., Richmond T.A., Molla M., Singh J., Green R.D., Berg D.E., Dailidiene D., Dailide G., Norton J.E., Kalia A., Richmond T.A., Molla M., Singh J., Green R.D., Berg D.E., Dailide G., Norton J.E., Kalia A., Richmond T.A., Molla M., Singh J., Green R.D., Berg D.E., Norton J.E., Kalia A., Richmond T.A., Molla M., Singh J., Green R.D., Berg D.E., Kalia A., Richmond T.A., Molla M., Singh J., Green R.D., Berg D.E., Richmond T.A., Molla M., Singh J., Green R.D., Berg D.E., Molla M., Singh J., Green R.D., Berg D.E., Singh J., Green R.D., Berg D.E., Green R.D., Berg D.E., Berg D.E. Mutation discovery in bacterial genomes: Metronidazole resistance in Helicobacter pylori. Nat. Methods. 2005;2:951–953. - PubMed
-
- Bandelt H., Dress A.W.M., Dress A.W.M. Split decomposition: A new and useful approach to phylogenetic analysis of distance data. Mol. Phylogenet. Evol. 1992;1:242–252. - PubMed
-
- Bitzan M., Ludwig K., Klemt M., Konig H., Buren J., Muller-Wiefel D.E., Ludwig K., Klemt M., Konig H., Buren J., Muller-Wiefel D.E., Klemt M., Konig H., Buren J., Muller-Wiefel D.E., Konig H., Buren J., Muller-Wiefel D.E., Buren J., Muller-Wiefel D.E., Muller-Wiefel D.E. The role of Escherichia coli O157 infections in the classical (enteropathic) hemolytic uraemic syndrome: Results of a central European, multicentre study. Epidemiol. Infect. 1993;110:183–196. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical