

Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function
- PMID: 16614756
- PMCID: PMC1435722
- DOI: 10.1172/JCI27540
Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function
Abstract
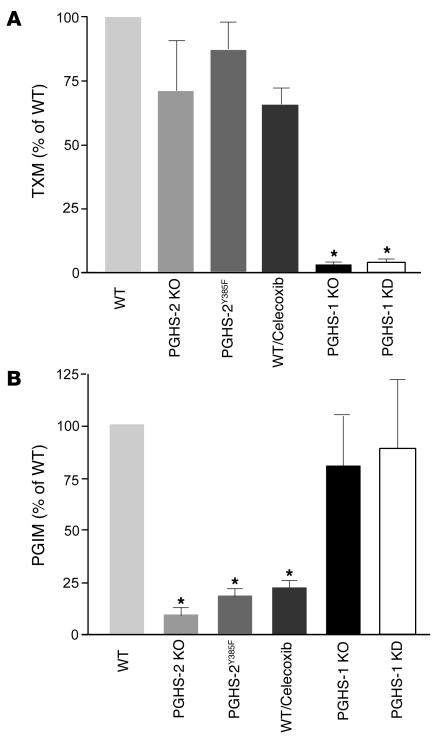
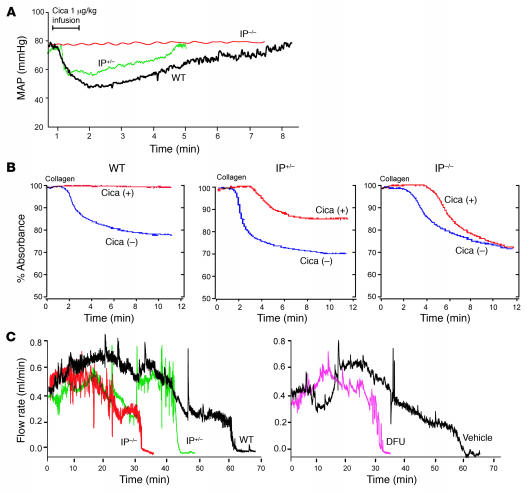
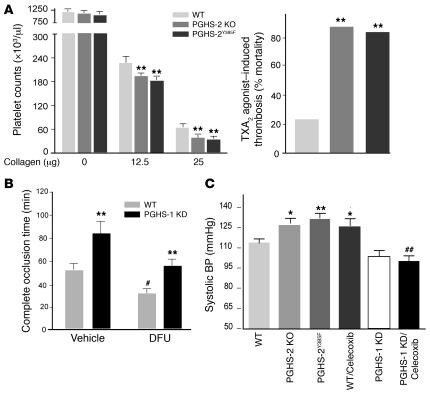
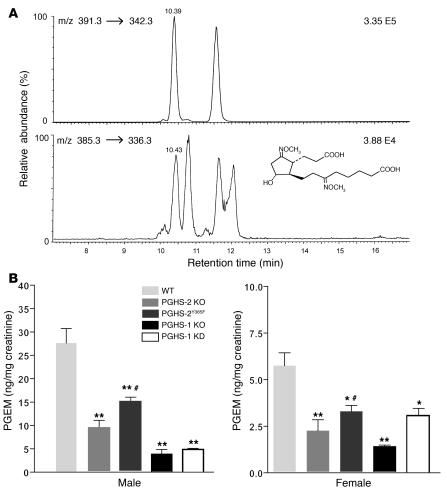
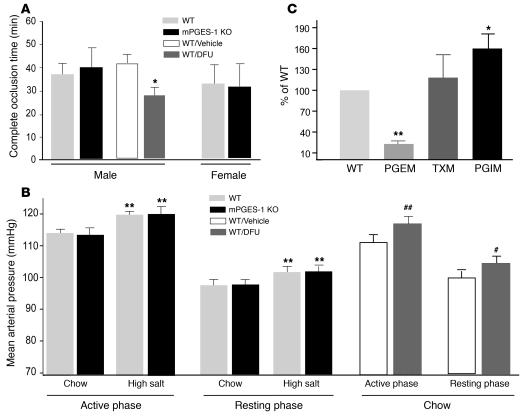
We investigated the mechanisms by which inhibitors of prostaglandin G/H synthase-2 (PGHS-2; known colloquially as COX-2) increase the incidence of myocardial infarction and stroke. These inhibitors are believed to exert both their beneficial and their adverse effects by suppression of PGHS-2-derived prostacyclin (PGI(2)) and PGE(2). Therefore, the challenge remains to identify a mechanism whereby PGI(2) and PGE(2) expression can be suppressed while avoiding adverse cardiovascular events. Here, selective inhibition, knockout, or mutation of PGHS-2, or deletion of the receptor for PGHS-2-derived PGI(2), was shown to accelerate thrombogenesis and elevate blood pressure in mice. These responses were attenuated by COX-1 knock down, which mimics the beneficial effects of low-dose aspirin. PGE(2) biosynthesis is catalyzed by the coordinate actions of COX enzymes and microsomal PGE synthase-1 (mPGES-1). We show that deletion of mPGES-1 depressed PGE(2) expression, augmented PGI(2) expression, and had no effect on thromboxane biosynthesis in vivo. Most importantly, mPGES-1 deletion affected neither thrombogenesis nor blood pressure. These results suggest that inhibitors of mPGES-1 may retain their antiinflammatory efficacy by depressing PGE(2), while avoiding the adverse cardiovascular consequences associated with PGHS-2-mediated PGI(2) suppression.
Figures
References
-
- Patrignani P., et al. COX-2 is not involved in thromboxane biosynthesis by activated human platelets. J. Physiol. Pharmacol. 1999;50:661–667. - PubMed
-
- Cheng Y., et al. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296:539–541. - PubMed
-
- FitzGerald G.A. Coxibs and cardiovascular disease. N. Engl. J. Med. 2004;351:1709–1711. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
