

Platelets secrete stromal cell-derived factor 1alpha and recruit bone marrow-derived progenitor cells to arterial thrombi in vivo
- PMID: 16618794
- PMCID: PMC2121205
- DOI: 10.1084/jem.20051772
Platelets secrete stromal cell-derived factor 1alpha and recruit bone marrow-derived progenitor cells to arterial thrombi in vivo
Abstract
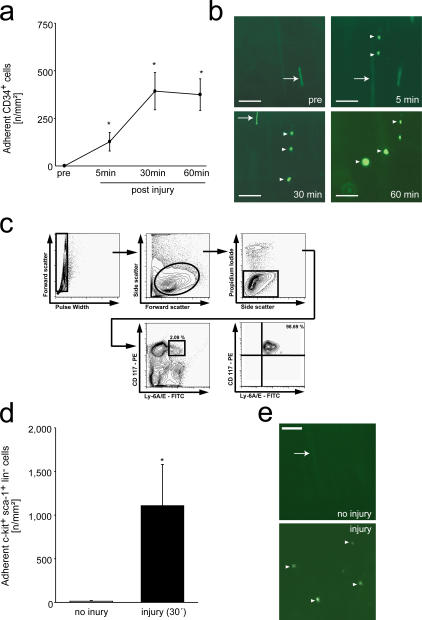
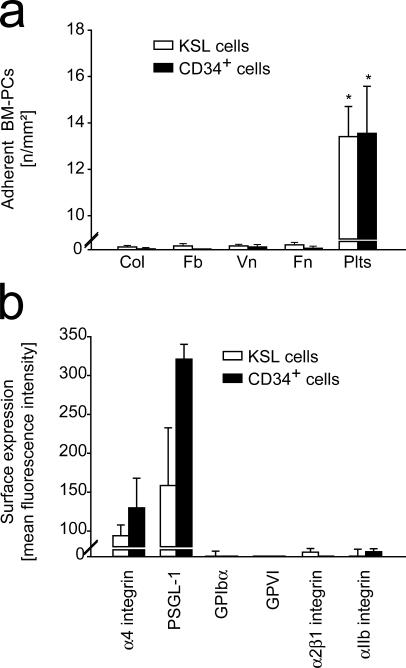
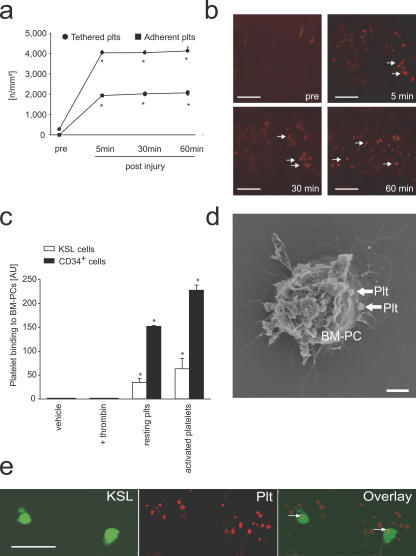
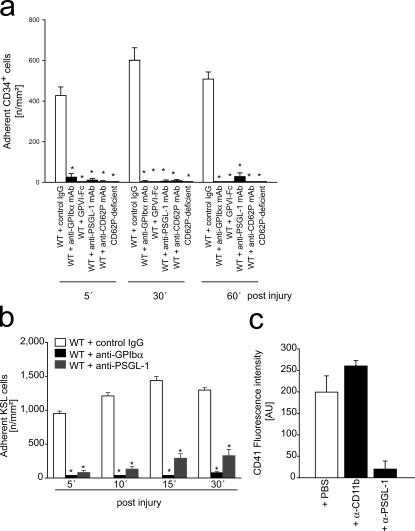
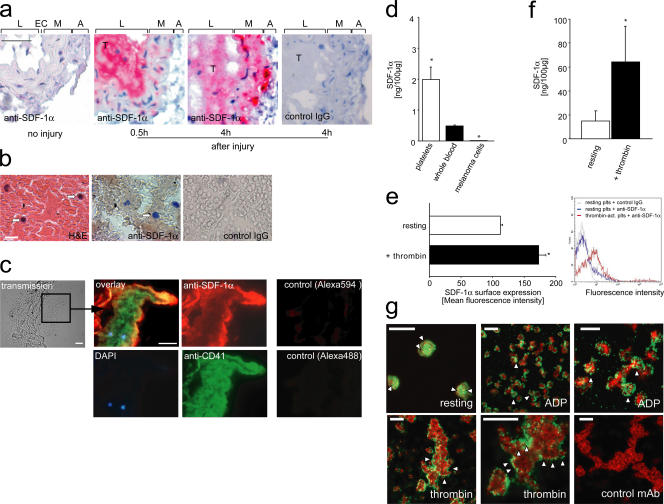
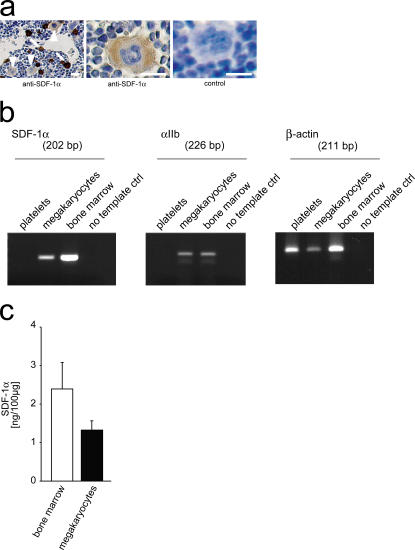
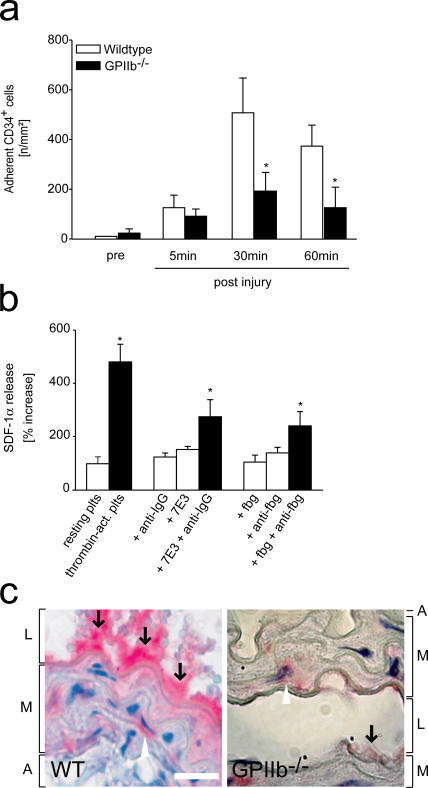
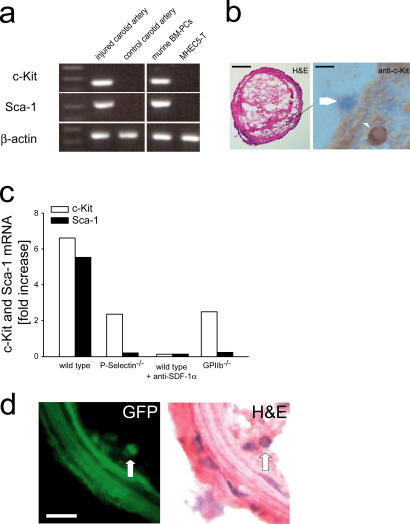
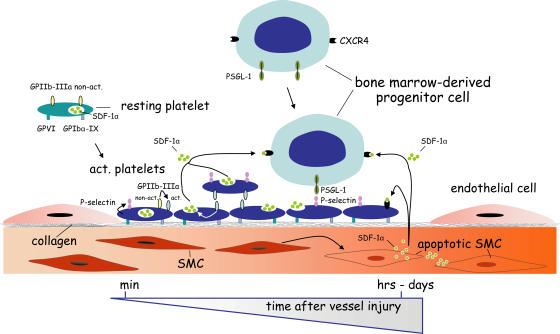
The accumulation of smooth muscle and endothelial cells is essential for remodeling and repair of injured blood vessel walls. Bone marrow-derived progenitor cells have been implicated in vascular repair and remodeling; however, the mechanisms underlying their recruitment to the site of injury remain elusive. Here, using real-time in vivo fluorescence microscopy, we show that platelets provide the critical signal that recruits CD34+ bone marrow cells and c-Kit+ Sca-1+ Lin- bone marrow-derived progenitor cells to sites of vascular injury. Correspondingly, specific inhibition of platelet adhesion virtually abrogated the accumulation of both CD34+ and c-Kit+ Sca-1+ Lin- bone marrow-derived progenitor cells at sites of endothelial disruption. Binding of bone marrow cells to platelets involves both P-selectin and GPIIb integrin on platelets. Unexpectedly, we found that activated platelets secrete the chemokine SDF-1alpha, thereby supporting further primary adhesion and migration of progenitor cells. These findings establish the platelet as a major player in the initiation of vascular remodeling, a process of fundamental importance for vascular repair and pathological remodeling after vascular injury.
Figures
Similar articles
-
SDF-1alpha/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells.Circ Res. 2005 Apr 15;96(7):784-91. doi: 10.1161/01.RES.0000162100.52009.38. Epub 2005 Mar 10. Circ Res. 2005. PMID: 15761195
-
Platelet-derived microparticles bind to hematopoietic stem/progenitor cells and enhance their engraftment.Blood. 2001 Nov 15;98(10):3143-9. doi: 10.1182/blood.v98.10.3143. Blood. 2001. PMID: 11698303
-
Platelets and stromal cell-derived factor-1 in progenitor cell recruitment.Semin Thromb Hemost. 2007 Mar;33(2):159-64. doi: 10.1055/s-2007-969029. Semin Thromb Hemost. 2007. PMID: 17340464 Review.
-
Platelet-derived stromal cell-derived factor-1 regulates adhesion and promotes differentiation of human CD34+ cells to endothelial progenitor cells.Circulation. 2008 Jan 15;117(2):206-15. doi: 10.1161/CIRCULATIONAHA.107.714691. Epub 2007 Dec 17. Circulation. 2008. PMID: 18086932
-
The role of progenitor cells in the development of intimal hyperplasia.J Vasc Surg. 2009 Feb;49(2):502-10. doi: 10.1016/j.jvs.2008.07.060. Epub 2008 Oct 22. J Vasc Surg. 2009. PMID: 18945574 Free PMC article. Review.
Cited by
-
A new platelet cryoprecipitate glue promoting bone formation after ectopic mesenchymal stromal cell-loaded biomaterial implantation in nude mice.Stem Cell Res Ther. 2013 Jan 4;4(1):1. doi: 10.1186/scrt149. Stem Cell Res Ther. 2013. PMID: 23290259 Free PMC article.
-
Lung megakaryocytes are immune modulatory cells.J Clin Invest. 2021 Jan 4;131(1):e137377. doi: 10.1172/JCI137377. J Clin Invest. 2021. PMID: 33079726 Free PMC article.
-
Enhanced mobilization of the bone marrow-derived circulating progenitor cells by intracoronary freshly isolated bone marrow cells transplantation in patients with acute myocardial infarction.J Cell Mol Med. 2012 Apr;16(4):852-64. doi: 10.1111/j.1582-4934.2011.01358.x. J Cell Mol Med. 2012. PMID: 21707914 Free PMC article. Clinical Trial.
-
Lactate stimulates vasculogenic stem cells via the thioredoxin system and engages an autocrine activation loop involving hypoxia-inducible factor 1.Mol Cell Biol. 2008 Oct;28(20):6248-61. doi: 10.1128/MCB.00795-08. Epub 2008 Aug 18. Mol Cell Biol. 2008. PMID: 18710947 Free PMC article.
-
Role of Stromal Cell-Derived Factor-1 in Endothelial Progenitor Cell-Mediated Vascular Repair and Regeneration.Tissue Eng Regen Med. 2021 Oct;18(5):747-758. doi: 10.1007/s13770-021-00366-9. Epub 2021 Aug 27. Tissue Eng Regen Med. 2021. PMID: 34449064 Free PMC article. Review.
References
-
- Carmeliet, P. 2000. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6:389–395. - PubMed
-
- Schwartz, S.M. 1997. Smooth muscle migration in atherosclerosis and restenosis. J. Clin. Invest. 100:S87–S89. - PubMed
-
- Werner, N., J. Priller, U. Laufs, M. Endres, M. Bohm, U. Dirnagl, and G. Nickenig. 2002. Bone marrow-derived progenitor cells modulate vascular reendothelialization and neointimal formation: effect of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition. Arterioscler. Thromb. Vasc. Biol. 22:1567–1572. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
