

Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models
- PMID: 16624935
- PMCID: PMC6673995
- DOI: 10.1523/JNEUROSCI.5461-05.2006
Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models
Abstract
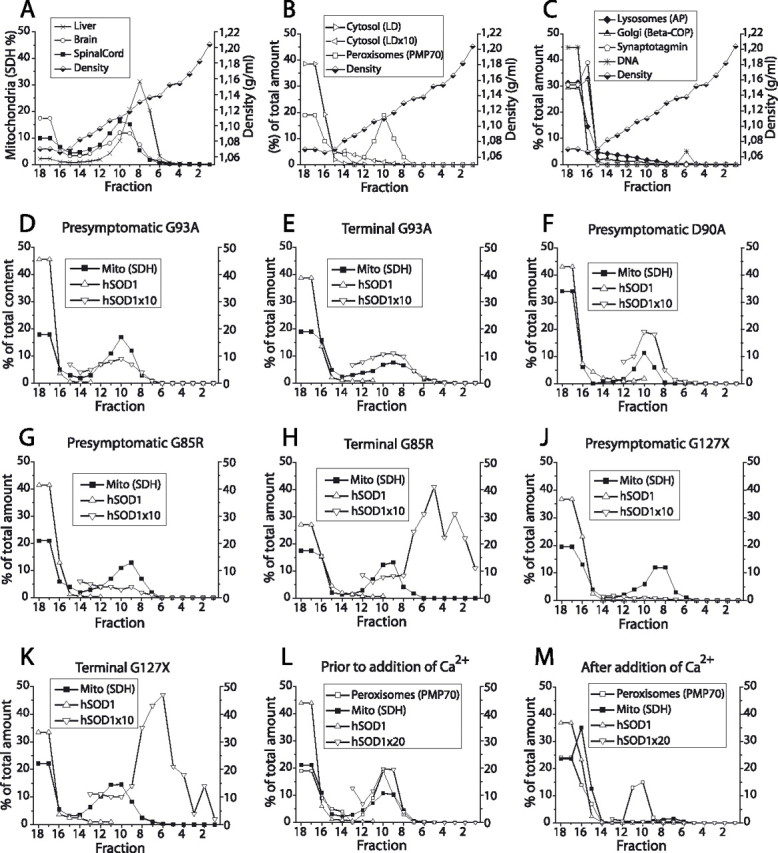
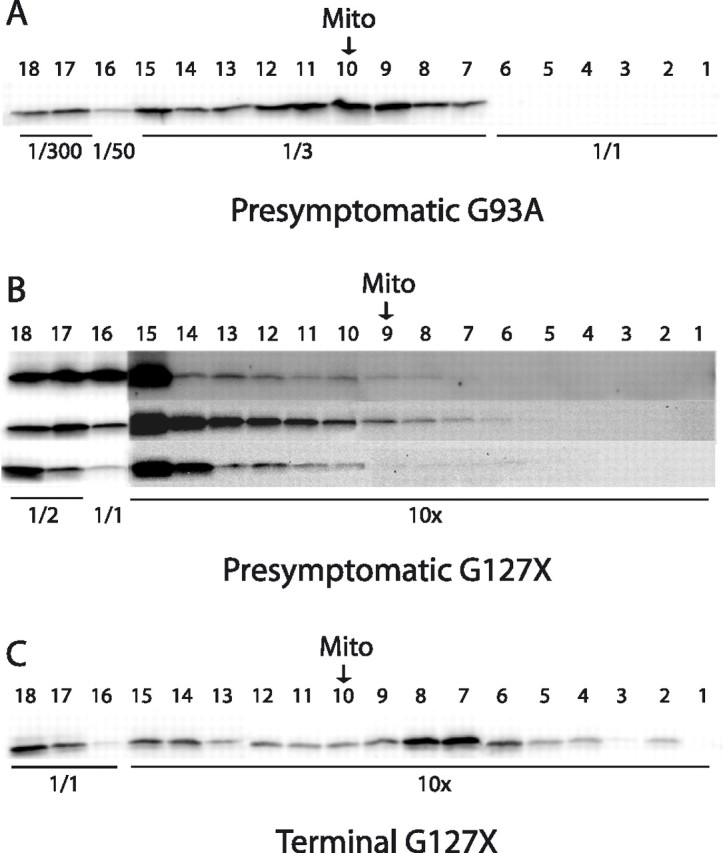
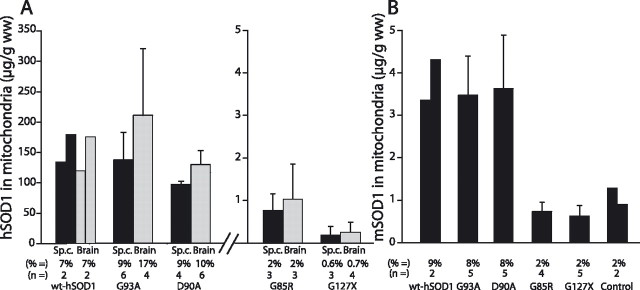
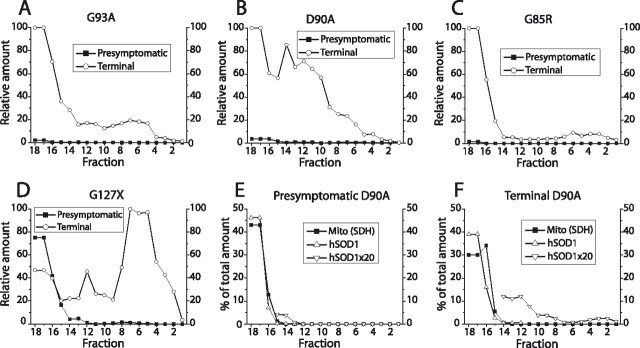
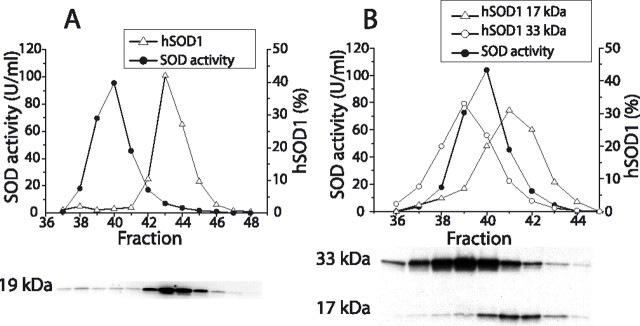
Mutants of human superoxide dismutase-1 (hSOD1) cause amyotrophic lateral sclerosis (ALS), and mitochondria are thought to be primary targets of the cytotoxic action. The high expression rates of hSOD1s in transgenic ALS models give high levels of the stable mutants G93A and D90A as well as the wild-type human enzyme, significant proportions of which lack Cu and the intrasubunit disulfide bond. The endogenous murine SOD1 (mSOD1) also lacks Cu and is disulfide reduced but is active and oxidized in mice expressing the low-level unstable mutants G85R and G127insTGGG. The possibility that the molecular alterations may cause artificial loading of the stable hSOD1s into mitochondria was explored. Approximately 10% of these hSOD1s were localized to mitochondria, reaching levels 100-fold higher than those of mSOD1 in control mice. There was no difference between brain and spinal cord and between stable mutants and the wild-type hSOD1. mSOD1 was increased fourfold in mitochondria from high-level hSOD1 mice but was normal in those with low levels, suggesting that the Cu deficiency and disulfide reduction cause mitochondrial overloading. The levels of G85R and G127insTGGG mutant hSOD1s in mitochondria were 100- and 1000-fold lower than those of stable mutants. Spinal cords from symptomatic mice contained hSOD1 aggregates covering the entire density gradient, which could contaminate isolated organelle fractions. Thus, high hSOD1 expression rates can cause artificial loading of mitochondria. Unstable low-level hSOD1s are excluded from mitochondria, indicating other primary locations of injury. Such models may be preferable for studies of ALS pathogenesis.
Figures
References
-
- Andersen PM, Nilsson P, Ala-Hurula V, Keranen ML, Tarvainen I, Haltia T, Nilsson L, Binzer M, Forsgren L, Marklund SL (1995). Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat Genet 10:61–66. - PubMed
-
- Andersen PM, Sims KB, Xin WW, Kiely R, O'Neill G, Ravits J, Pioro E, Harati Y, Brower RD, Levine JS, Heinicke HU, Seltzer W, Boss M, Brown RH Jr (2003). Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord 2:62–73. - PubMed
-
- Bowling AC, Schulz JB, Brown RH Jr, Beal MF (1993). Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem 61:2322–2325. - PubMed
-
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW (1997). ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18:327–338. - PubMed
-
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW (1998). Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281:1851–1854. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous