

Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system
- PMID: 16624948
- PMCID: PMC6673997
- DOI: 10.1523/JNEUROSCI.5078-05.2006
Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system
Abstract
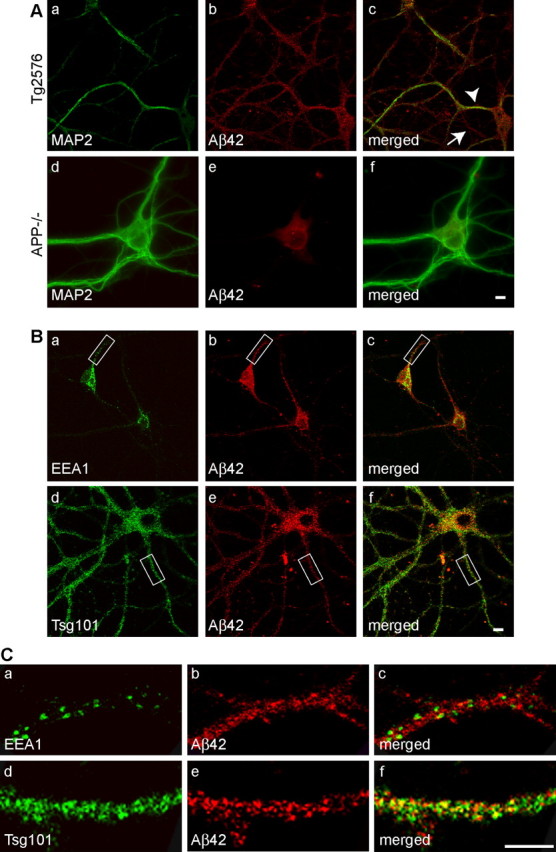
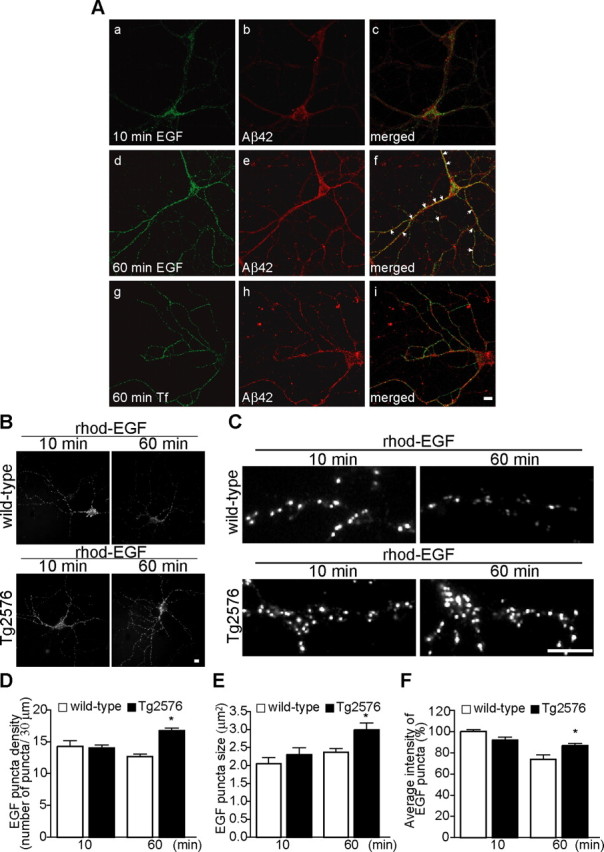
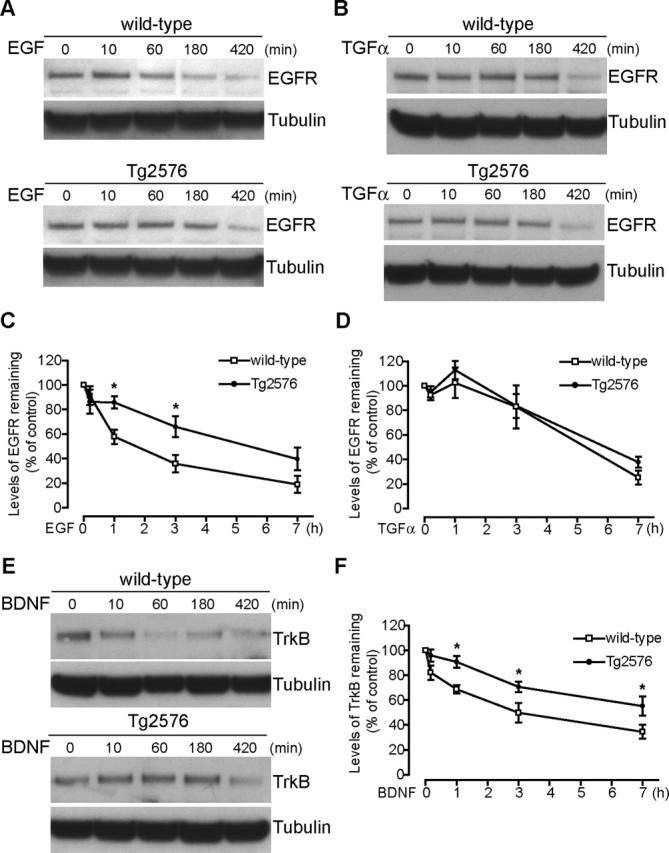
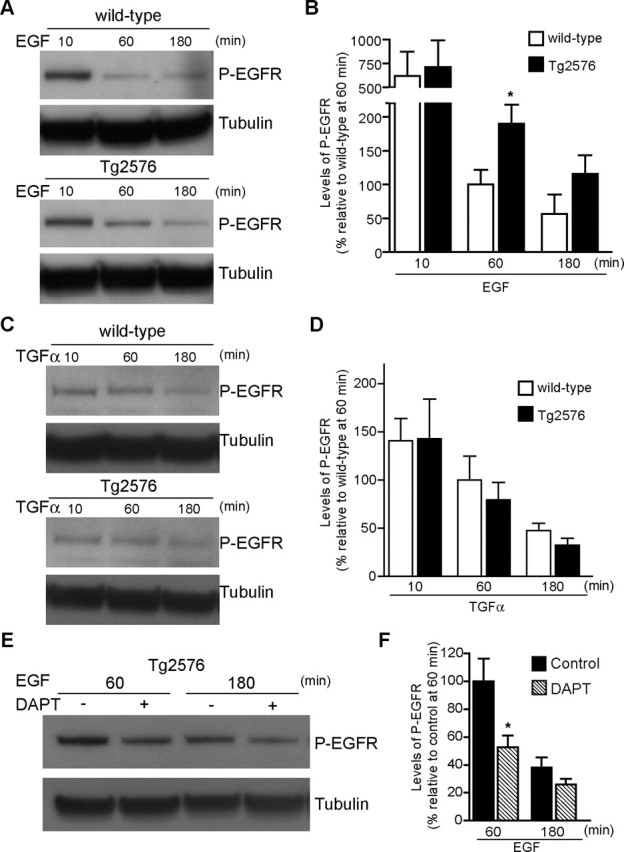
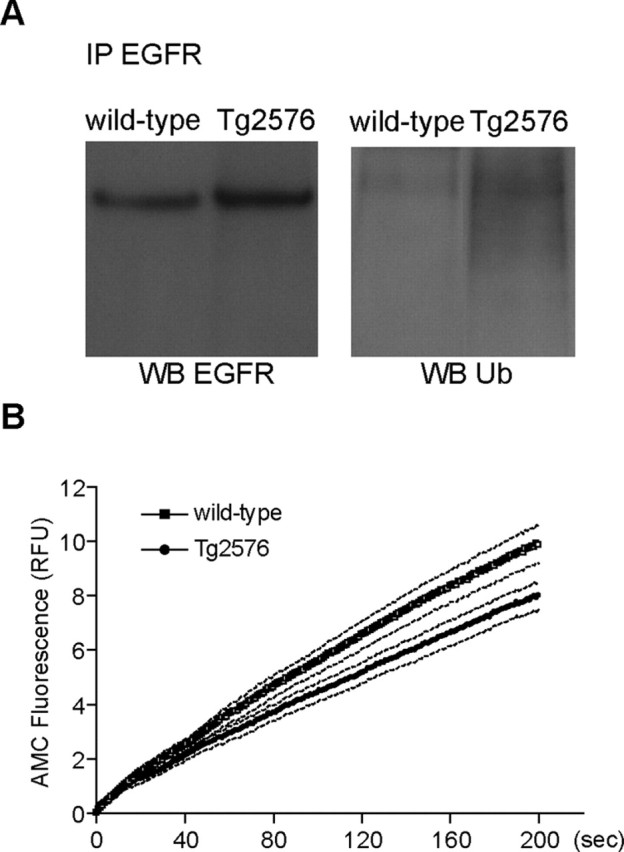
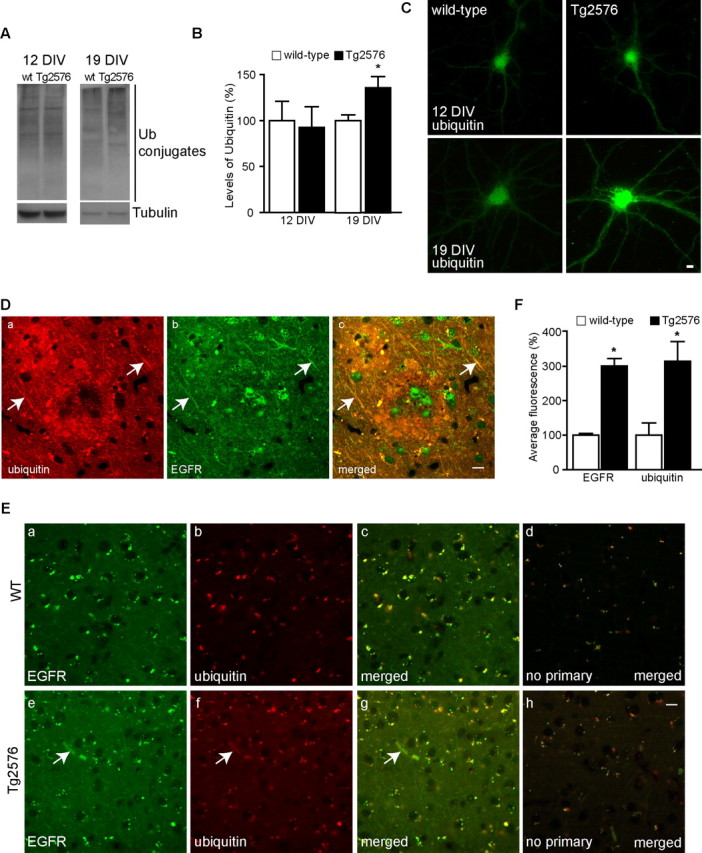
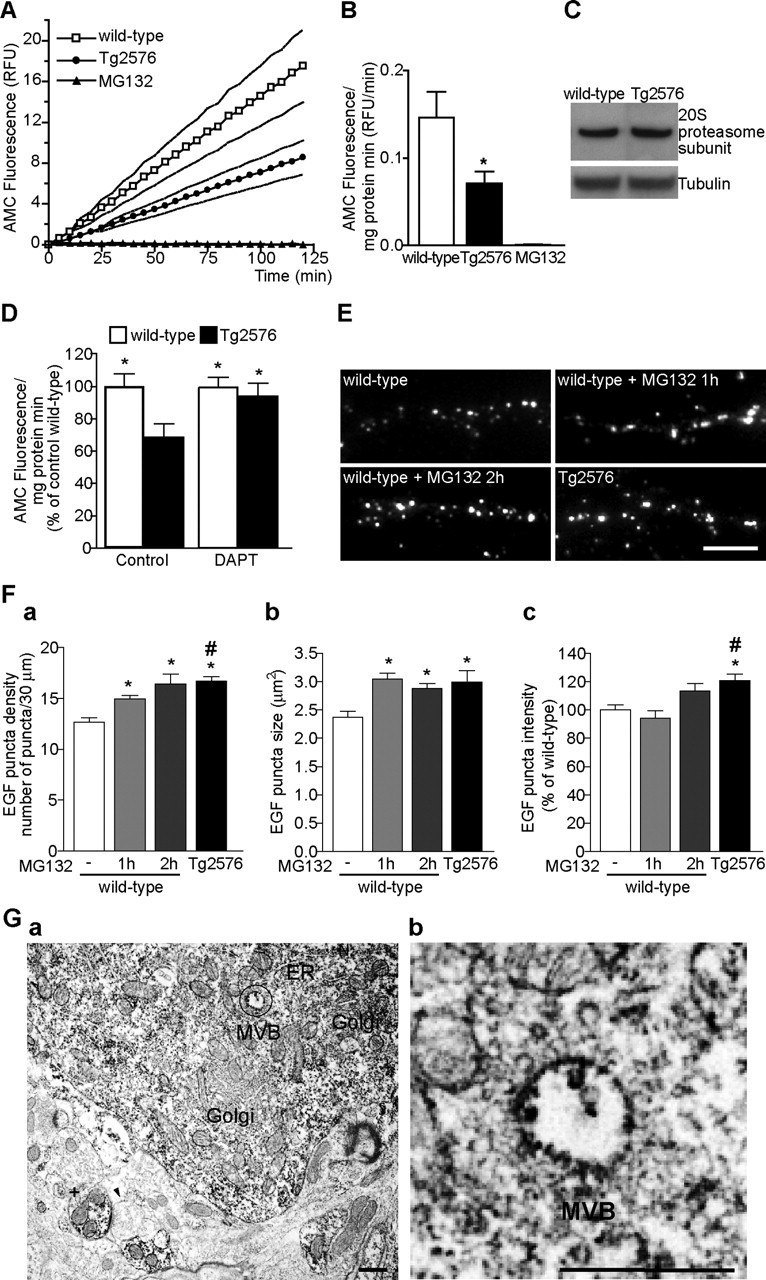
Increasing evidence links intraneuronal beta-amyloid (Abeta42) accumulation with the pathogenesis of Alzheimer's disease (AD). In Abeta precursor protein (APP) mutant transgenic mice and in human AD brain, progressive intraneuronal accumulation of Abeta42 occurs especially in multivesicular bodies (MVBs). We hypothesized that this impairs the MVB sorting pathway. We used the trafficking of the epidermal growth factor receptor (EGFR) and TrkB receptor to investigate the MVB sorting pathway in cultured neurons. We report that, during EGF stimulation, APP mutant neurons demonstrated impaired inactivation, degradation, and ubiquitination of EGFR. EGFR degradation is dependent on translocation from MVB outer to inner membranes, which is regulated by the ubiquitin-proteasome system (UPS). We provide evidence that Abeta accumulation in APP mutant neurons inhibits the activities of the proteasome and deubiquitinating enzymes. These data suggest a mechanism whereby Abeta accumulation in neurons impairs the MVB sorting pathway via the UPS in AD.
Figures
References
-
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK (2005). Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis 20:187–198. - PubMed
-
- Alwan HA, van Zoelen EJ, van Leeuwen JE (2003). Ligand-induced lysosomal epidermal growth factor receptor (EGFR) degradation is preceded by proteasome-dependent EGFR de-ubiquitination. J Biol Chem 278:35781–35790. - PubMed
-
- Babst M (2005). A protein’s final ESCRT. Traffic 6:2–9. - PubMed
-
- Bertram L, Hiltunen M, Parkinson M, Ingelsson M, Lange C, Ramasamy K, Mullin K, Menon R, Sampson AJ, Hsiao MY, Elliott KJ, Velicelebi G, Moscarillo T, Hyman BT, Wagner SL, Becker KD, Blacker D, Tanzi RE (2005). Family-based association between Alzheimer’s disease and variants in UBQLN1. N Engl J Med 352:884–894. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous