

Barrel map development relies on protein kinase A regulatory subunit II beta-mediated cAMP signaling
- PMID: 16624954
- PMCID: PMC6674004
- DOI: 10.1523/JNEUROSCI.3745-05.2006
Barrel map development relies on protein kinase A regulatory subunit II beta-mediated cAMP signaling
Abstract
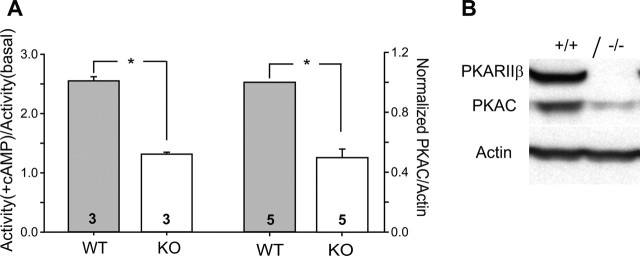
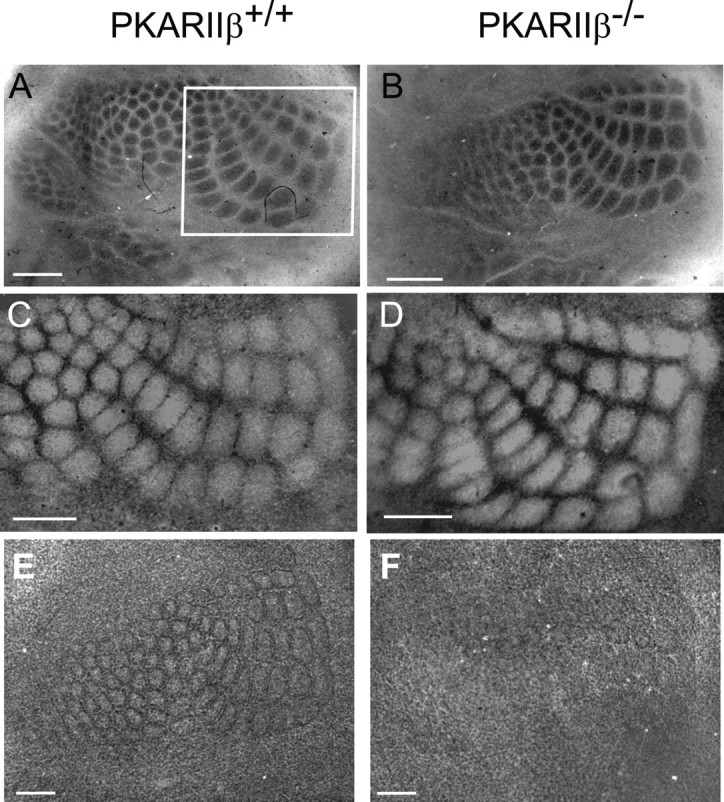
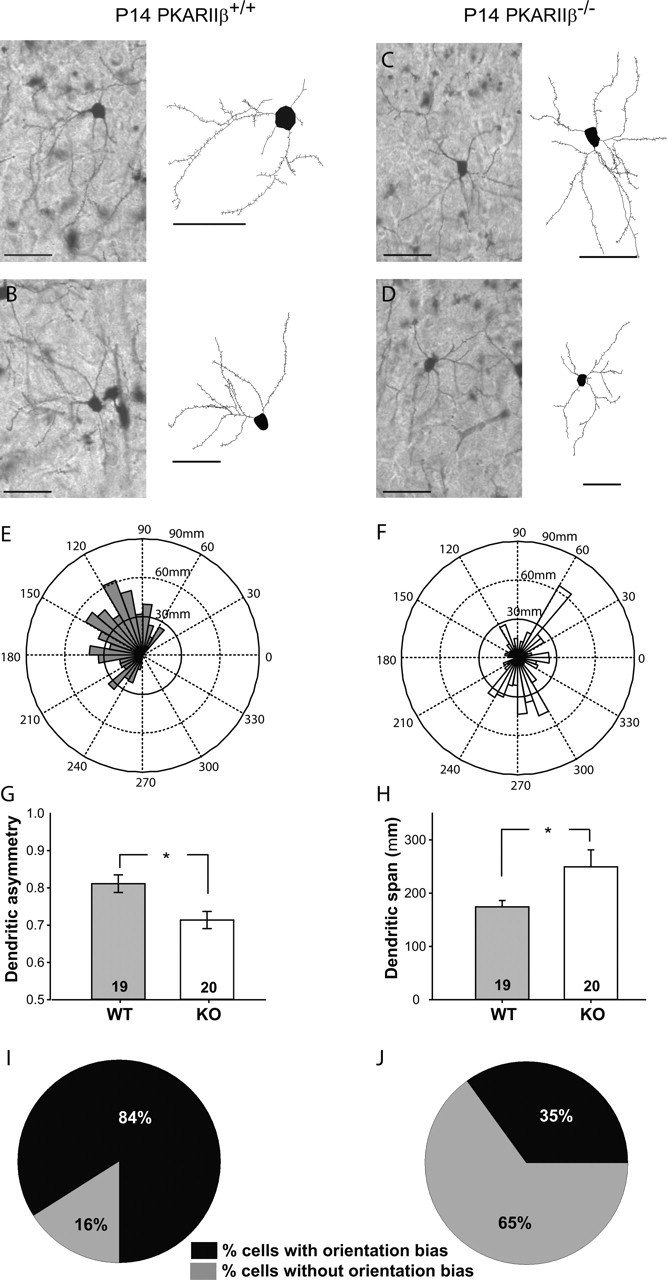
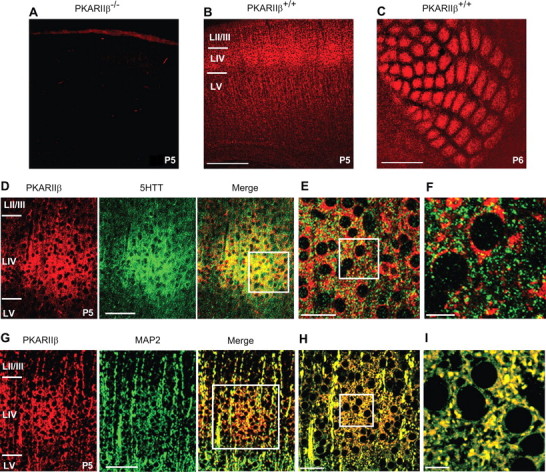
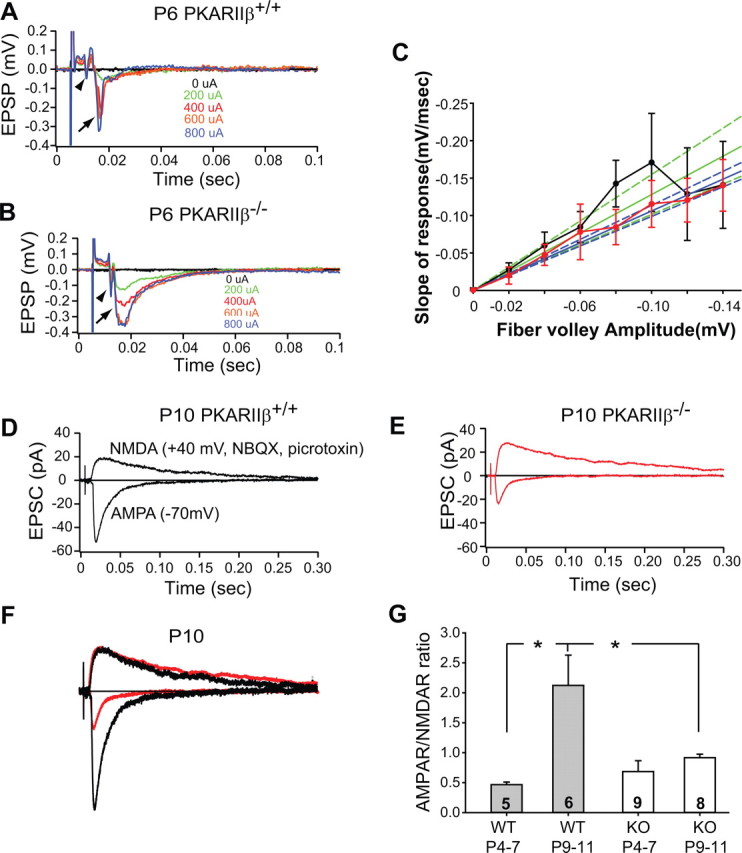
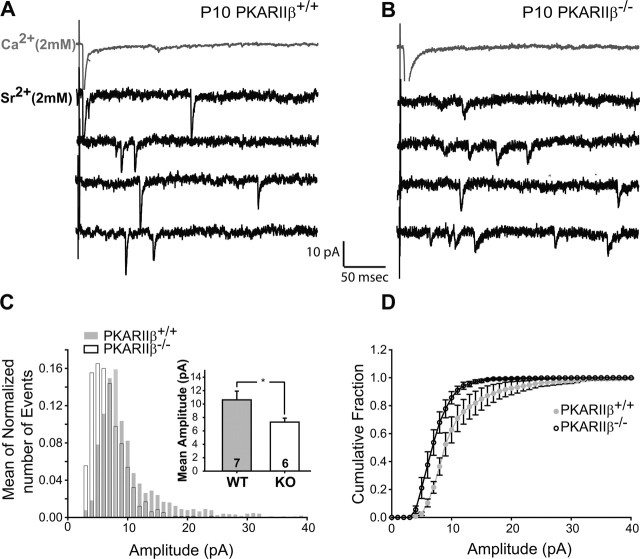
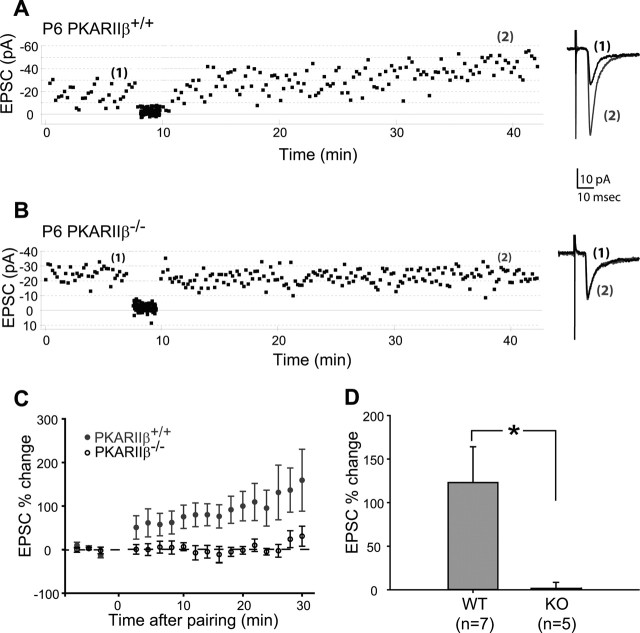
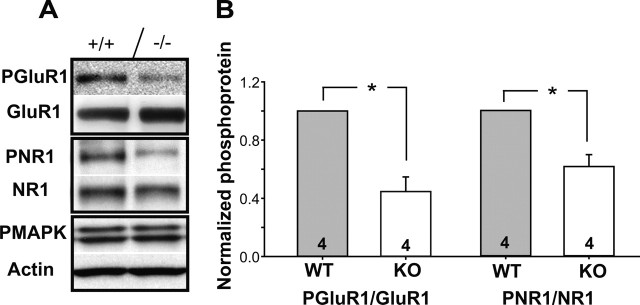
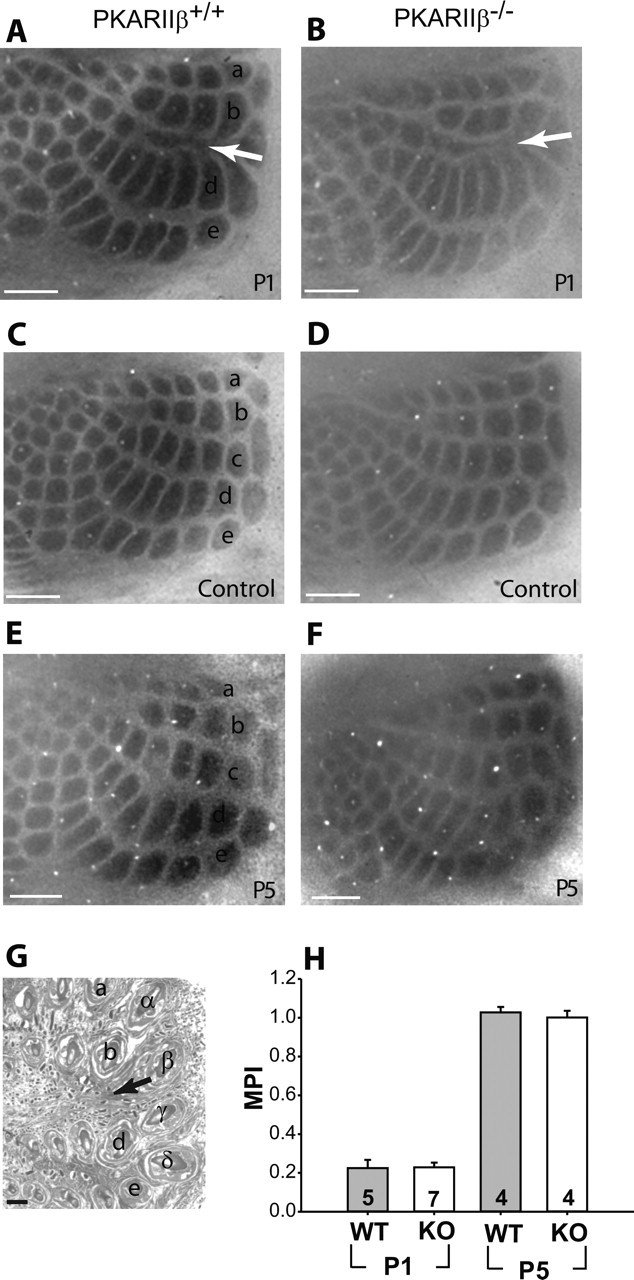
The cellular and molecular mechanisms mediating the activity-dependent development of brain circuitry are still incompletely understood. Here, we examine the role of cAMP-dependent protein kinase [protein kinase A (PKA)] signaling in cortical development and plasticity, focusing on its role in thalamocortical synapse and barrel map development. We provide direct evidence that PKA activity mediates barrel map formation using knock-out mice that lack type IIbeta regulatory subunits of PKA (PKARIIbeta). We show that PKARIIbeta-mediated PKA function is required for proper dendritogenesis and the organization of cortical layer IV neurons into barrels, but not for the development and plasticity of thalamocortical afferent clustering into a barrel pattern. We localize PKARIIbeta function to postsynaptic processes in barrel cortex and show that postsynaptic PKA targets, but not presynaptic PKA targets, have decreased phosphorylation in pkar2b knock-out (PKARIIbeta(-/-)) mice. We also show that long-term potentiation at TC synapses and the associated developmental increase in AMPA receptor function at these synapses, which normally occurs as barrels form, is absent in PKARIIbeta(-/-) mice. Together, these experiments support an activity-dependent model for barrel map development in which the selective addition and elimination of thalamocortical synapses based on Hebbian mechanisms for synapse formation is mediated by a cAMP/PKA-dependent pathway that relies on PKARIIbeta function.
Figures
Similar articles
-
Cortical adenylyl cyclase 1 is required for thalamocortical synapse maturation and aspects of layer IV barrel development.J Neurosci. 2008 Jun 4;28(23):5931-43. doi: 10.1523/JNEUROSCI.0815-08.2008. J Neurosci. 2008. PMID: 18524897 Free PMC article.
-
Involvement of protein kinase A in patterning of the mouse somatosensory cortex.J Neurosci. 2006 May 17;26(20):5393-401. doi: 10.1523/JNEUROSCI.0750-06.2006. J Neurosci. 2006. PMID: 16707791 Free PMC article.
-
Requirement for the RIIbeta isoform of PKA, but not calcium-stimulated adenylyl cyclase, in visual cortical plasticity.J Neurosci. 2004 Oct 13;24(41):9049-58. doi: 10.1523/JNEUROSCI.2409-04.2004. J Neurosci. 2004. PMID: 15483123 Free PMC article.
-
Regulation of neuronal PKA signaling through AKAP targeting dynamics.Eur J Cell Biol. 2006 Jul;85(7):627-33. doi: 10.1016/j.ejcb.2006.01.010. Epub 2006 Feb 28. Eur J Cell Biol. 2006. PMID: 16504338 Review.
-
The cAMP-dependent protein kinases and cAMP signal transduction.Semin Cancer Biol. 1994 Aug;5(4):285-94. Semin Cancer Biol. 1994. PMID: 7803765 Review.
Cited by
-
Systemic administration of rolipram increases medullary and spinal cAMP and activates a latent respiratory motor pathway after high cervical spinal cord injury.J Spinal Cord Med. 2009;32(2):175-82. doi: 10.1080/10790268.2009.11760769. J Spinal Cord Med. 2009. PMID: 19569465 Free PMC article.
-
Roles of mGluR5 in synaptic function and plasticity of the mouse thalamocortical pathway.Eur J Neurosci. 2009 Apr;29(7):1379-96. doi: 10.1111/j.1460-9568.2009.06696.x. Epub 2009 Mar 23. Eur J Neurosci. 2009. PMID: 19519626 Free PMC article.
-
In DRG11 knock-out mice, trigeminal cell death is extensive and does not account for failed brainstem patterning.J Neurosci. 2008 Apr 2;28(14):3577-85. doi: 10.1523/JNEUROSCI.4203-07.2008. J Neurosci. 2008. PMID: 18385316 Free PMC article.
-
Conditional Dnmt1 deletion in dorsal forebrain disrupts development of somatosensory barrel cortex and thalamocortical long-term potentiation.Thalamus Relat Syst. 2005 Sep;3(3):227-233. doi: 10.1017/S1472928807000222. Thalamus Relat Syst. 2005. PMID: 17710197 Free PMC article.
-
Genetically encoded sensors towards imaging cAMP and PKA activity in vivo.J Neurosci Methods. 2021 Oct 1;362:109298. doi: 10.1016/j.jneumeth.2021.109298. Epub 2021 Jul 31. J Neurosci Methods. 2021. PMID: 34339753 Free PMC article. Review.
References
-
- Abdel-Majid RM, Leong WL, Schalkwyk LC, Smallman DS, Wong ST, Storm DR, Fine A, Dobson MJ, Guernsey DL, Neumann PE (1998). Loss of adenylyl cyclase I activity disrupts patterning of mouse somatosensory cortex. Nat Genet 19:289–291. - PubMed
-
- Amieux PS, Cummings DE, Motamed K, Brandon EP, Wailes LA, Le K, Idzerda RL, McKnight GS (1997). Compensatory regulation of RIalpha protein levels in protein kinase A mutant mice. J Biol Chem 272:3993–3998. - PubMed
-
- Bauman AL, Goehring AS, Scott JD (2004). Orchestration of synaptic plasticity through AKAP signaling complexes. Neuropharmacology 46:299–310. - PubMed
-
- Beaver CJ, Ji Q, Fischer QS, Daw NW (2001). Cyclic AMP-dependent protein kinase mediates ocular dominance shifts in cat visual cortex. Nat Neurosci 4:159–163. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases