

Identification and classification of conserved RNA secondary structures in the human genome
- PMID: 16628248
- PMCID: PMC1440920
- DOI: 10.1371/journal.pcbi.0020033
Identification and classification of conserved RNA secondary structures in the human genome
Abstract
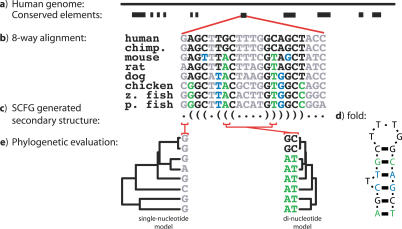
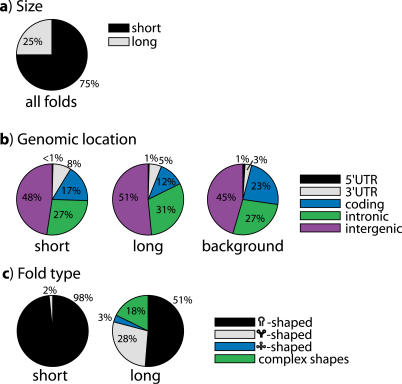
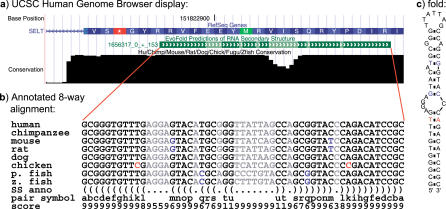
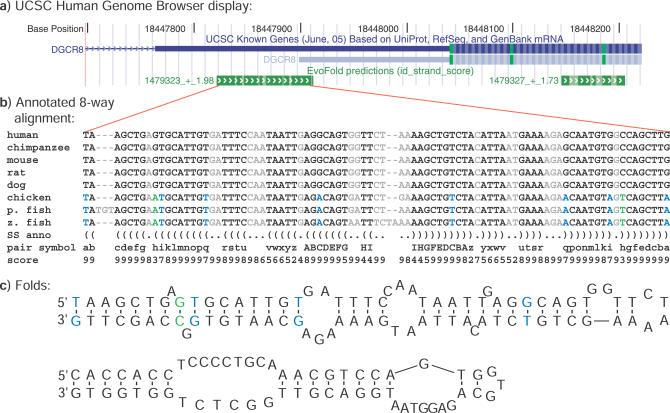
The discoveries of microRNAs and riboswitches, among others, have shown functional RNAs to be biologically more important and genomically more prevalent than previously anticipated. We have developed a general comparative genomics method based on phylogenetic stochastic context-free grammars for identifying functional RNAs encoded in the human genome and used it to survey an eight-way genome-wide alignment of the human, chimpanzee, mouse, rat, dog, chicken, zebra-fish, and puffer-fish genomes for deeply conserved functional RNAs. At a loose threshold for acceptance, this search resulted in a set of 48,479 candidate RNA structures. This screen finds a large number of known functional RNAs, including 195 miRNAs, 62 histone 3'UTR stem loops, and various types of known genetic recoding elements. Among the highest-scoring new predictions are 169 new miRNA candidates, as well as new candidate selenocysteine insertion sites, RNA editing hairpins, RNAs involved in transcript auto regulation, and many folds that form singletons or small functional RNA families of completely unknown function. While the rate of false positives in the overall set is difficult to estimate and is likely to be substantial, the results nevertheless provide evidence for many new human functional RNAs and present specific predictions to facilitate their further characterization.
Conflict of interest statement
Figures

References
-
- Eddy SR. Non-coding RNA genes and the modern RNA world. Nat Rev Genet. 2001;2:919–929. - PubMed
-
- Bompfünewerer AF, Flamm C, Fried C, Fritzsch G, Hofacker IL, et al. Evolutionary patterns of non-coding RNAs. Theor Biosci. 2004;123:301–369. - PubMed
-
- Mattick JS, Makunin IV. Small regulatory RNAs in mammals. Hum Mol Genet 14 Spec No. 2005;1:R121–R132. - PubMed
-
- Brosius J. The contribution of RNAs and retroposition to evolutionary novelties. Genetica. 2003;118:99–116. - PubMed
-
- Rivas E, Eddy SR. Secondary structure alone is generally not statistically significant for the detection of noncoding rnas. Bioinformatics. 2000;16:583–605. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
