

Cyclopentenone isoprostanes are novel bioactive products of lipid oxidation which enhance neurodegeneration
- PMID: 16638022
- PMCID: PMC2881557
- DOI: 10.1111/j.1471-4159.2006.03797.x
Cyclopentenone isoprostanes are novel bioactive products of lipid oxidation which enhance neurodegeneration
Abstract
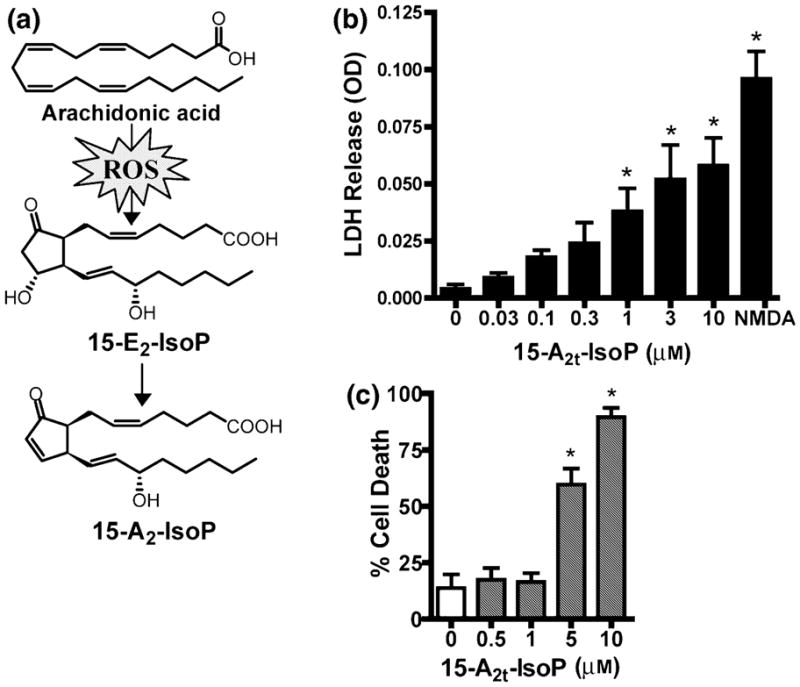
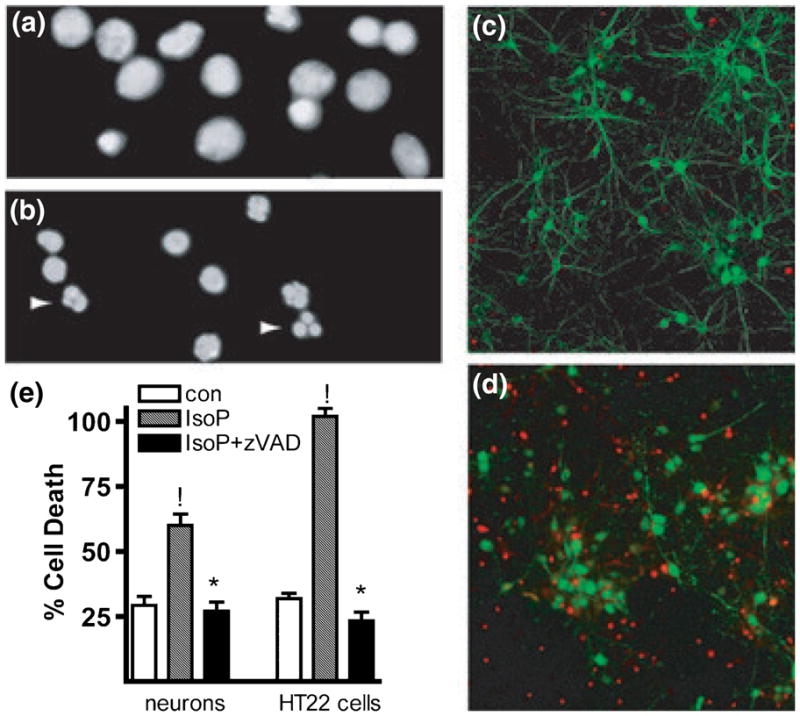
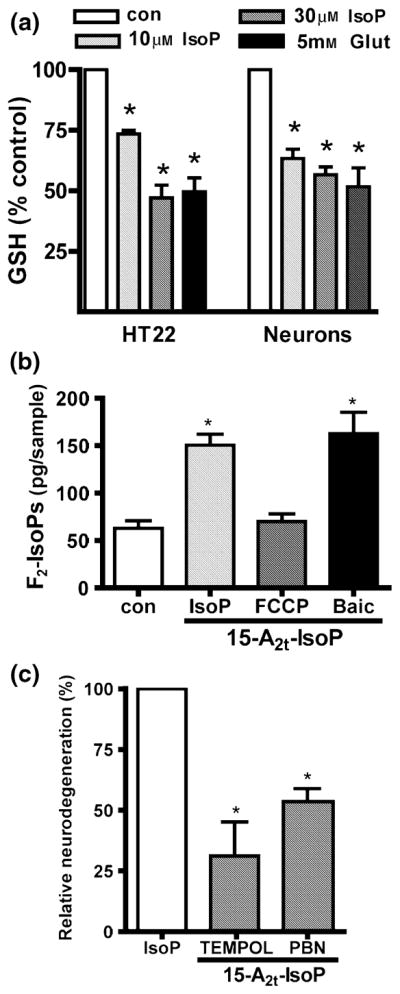
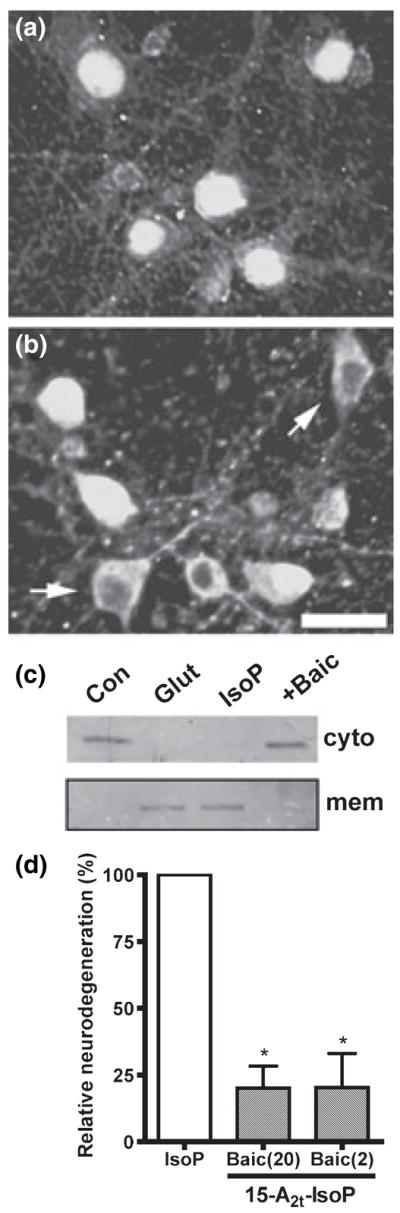
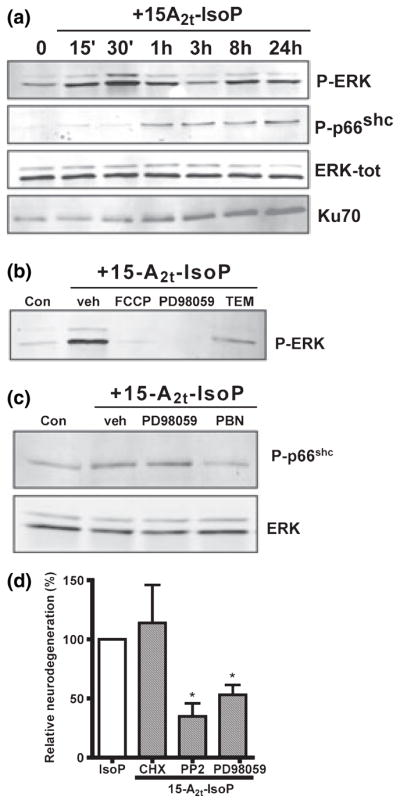
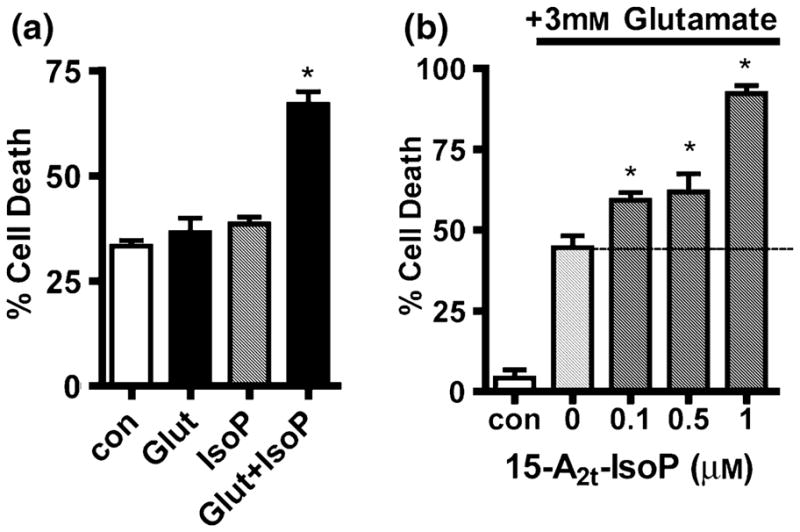
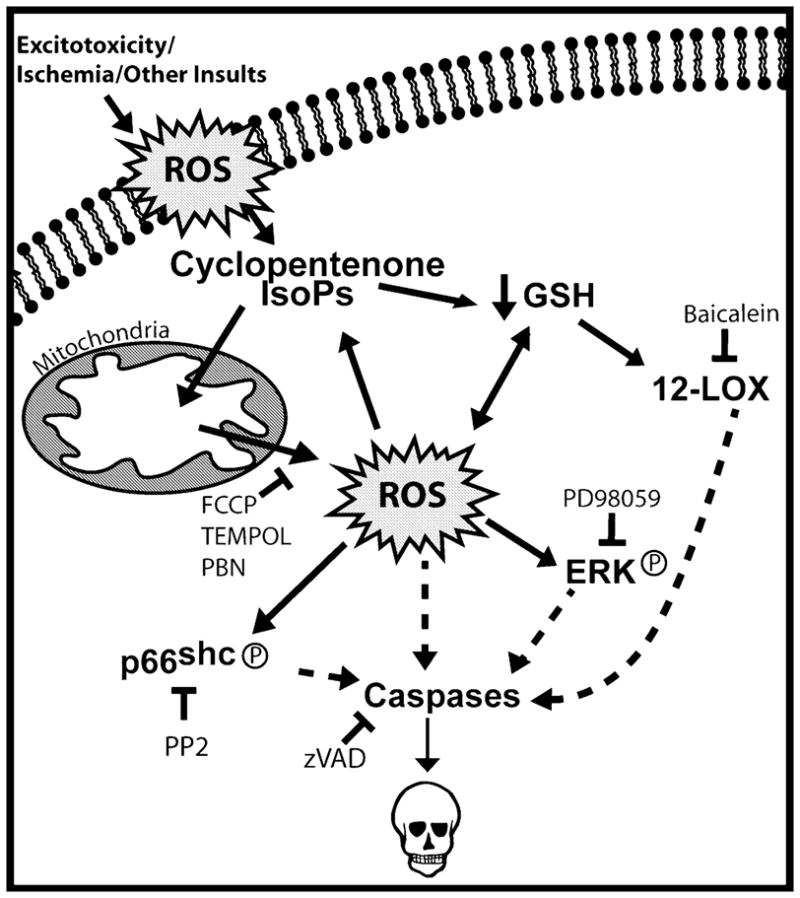
Oxidative stress and subsequent lipid peroxidation are involved in the pathogenesis of numerous neurodegenerative conditions, including stroke. Cyclopentenone isoprostanes (IsoPs) are novel electrophilic lipid peroxidation products formed under conditions of oxidative stress via the isoprostane pathway. These cyclopentenone IsoPs are isomeric to highly bioactive cyclopentenone prostaglandins, yet it has not been determined if these products are biologically active or are formed in the brain. Here we demonstrate that the major cyclopentenone IsoP isomer 15-A2t-IsoP potently induces apoptosis in neuronal cultures at submicromolar concentrations. We present a model in which 15-A2t-IsoP induced neuronal apoptosis involves initial depletion of glutathione and enhanced production of reactive oxygen species, followed by 12-lipoxygenase activation and phosphorylation of extracellular signal-regulated kinase 1/2 and the redox sensitive adaptor protein p66shc, which results in caspase-3 cleavage. 15-A2t-IsoP application also dramatically potentiates oxidative glutamate toxicity at concentrations as low as 100 nm, demonstrating the functional importance of these molecules in neurodegeneration. Finally, we employ novel mass spectrometric methods to show that cyclopentenone IsoPs are formed abundantly in brain tissue under conditions of oxidative stress. Together these findings suggest that cyclopentenone IsoPs may contribute to neuronal death caused by oxidative insults, and that their activity should perhaps be addressed when designing neuroprotective therapies.
Figures
References
-
- Arai K, Nishiyama N, Matsuki N, Ikegaya Y. Neuro-protective effects of lipoxygenase inhibitors against ischemic injury in rat hippocampal slice cultures. Brain Res. 2001;904:167–172. - PubMed
-
- Balamurugan K, Rajaram R, Ramasami T, Narayanan S. Chromium (III) – induced apoptosis of lymphocytes: death decision by ROS and Src-family tyrosine kinases. Free Radic Biol Med. 2002;33:1622–1640. - PubMed
-
- Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. - PubMed
-
- Bruckner SR, Perry G, Estus S. 4-hydroxynonenal contributes to NGF withdrawal-induced neuronal apoptosis. J Neurochem. 2003;85:999–1005. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
