

The genome of Rhizobium leguminosarum has recognizable core and accessory components
- PMID: 16640791
- PMCID: PMC1557990
- DOI: 10.1186/gb-2006-7-4-r34
The genome of Rhizobium leguminosarum has recognizable core and accessory components
Abstract
Background: Rhizobium leguminosarum is an alpha-proteobacterial N2-fixing symbiont of legumes that has been the subject of more than a thousand publications. Genes for the symbiotic interaction with plants are well studied, but the adaptations that allow survival and growth in the soil environment are poorly understood. We have sequenced the genome of R. leguminosarum biovar viciae strain 3841.
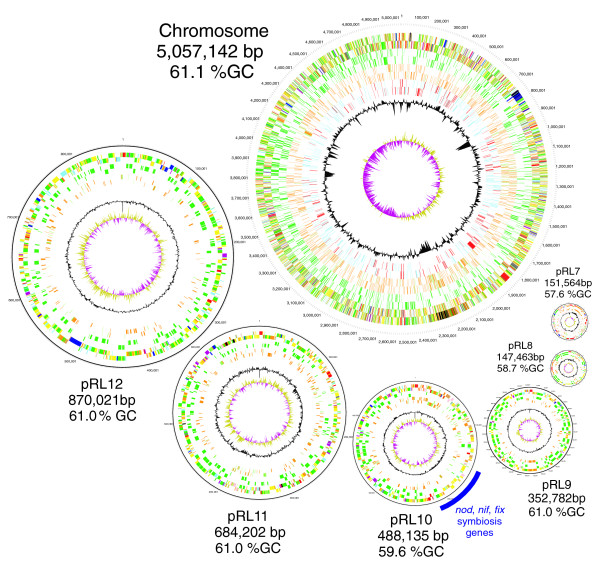
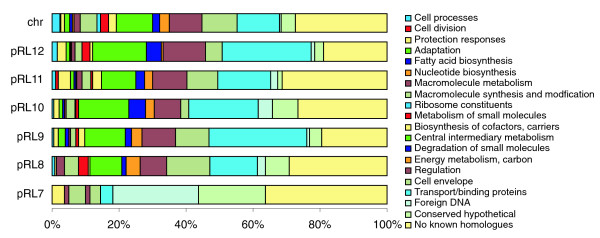
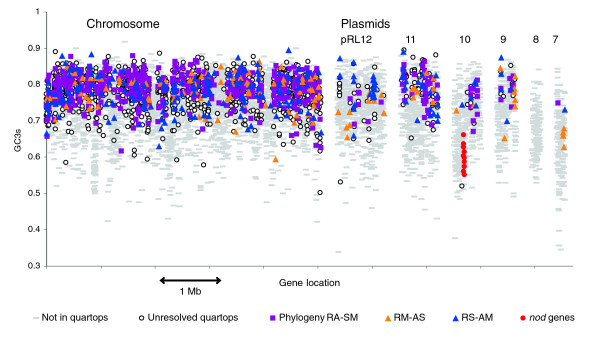
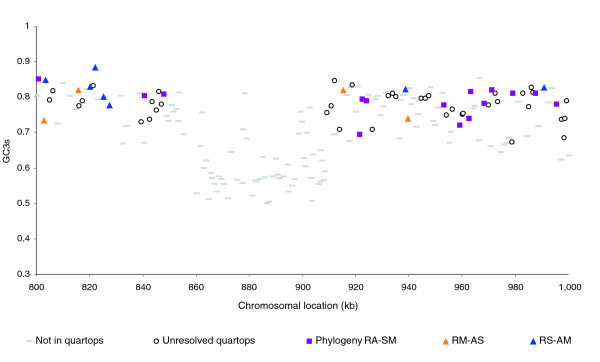
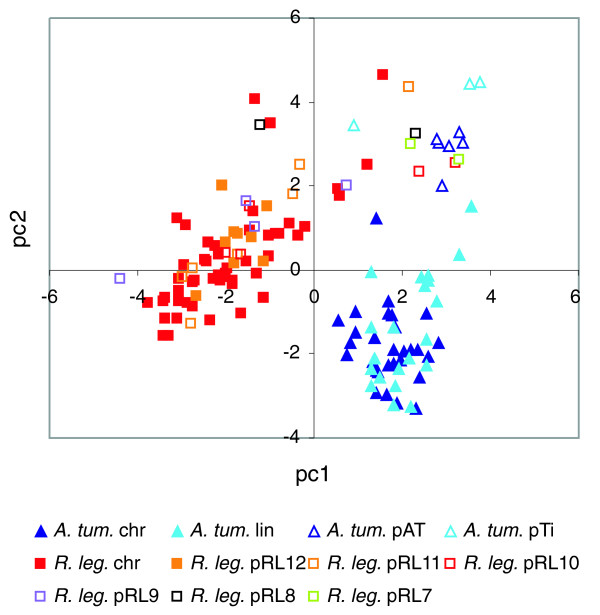
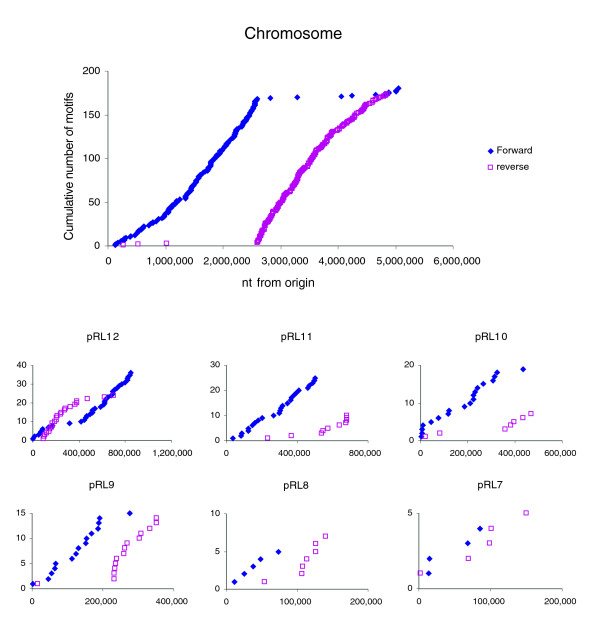
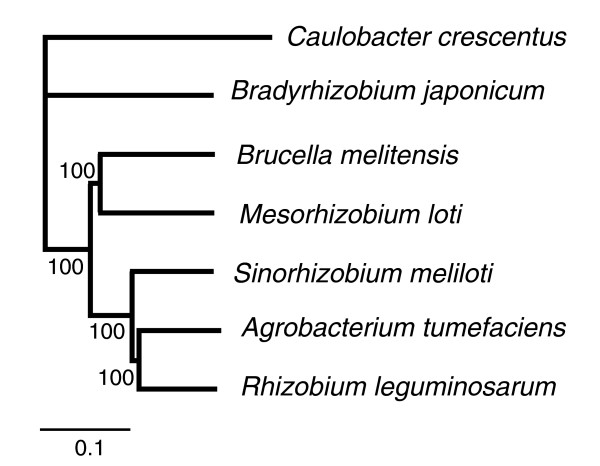
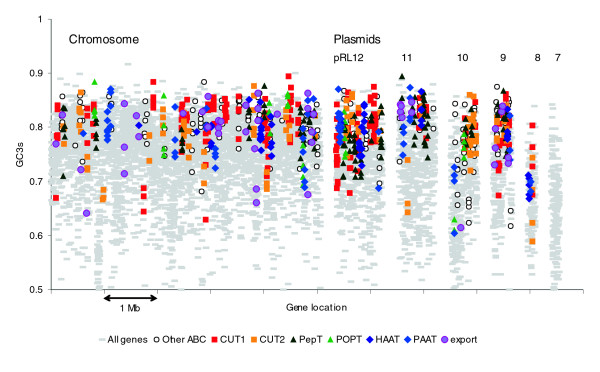
Results: The 7.75 Mb genome comprises a circular chromosome and six circular plasmids, with 61% G+C overall. All three rRNA operons and 52 tRNA genes are on the chromosome; essential protein-encoding genes are largely chromosomal, but most functional classes occur on plasmids as well. Of the 7,263 protein-encoding genes, 2,056 had orthologs in each of three related genomes (Agrobacterium tumefaciens, Sinorhizobium meliloti, and Mesorhizobium loti), and these genes were over-represented in the chromosome and had above average G+C. Most supported the rRNA-based phylogeny, confirming A. tumefaciens to be the closest among these relatives, but 347 genes were incompatible with this phylogeny; these were scattered throughout the genome but were over-represented on the plasmids. An unexpectedly large number of genes were shared by all three rhizobia but were missing from A. tumefaciens.
Conclusion: Overall, the genome can be considered to have two main components: a 'core', which is higher in G+C, is mostly chromosomal, is shared with related organisms, and has a consistent phylogeny; and an 'accessory' component, which is sporadic in distribution, lower in G+C, and located on the plasmids and chromosomal islands. The accessory genome has a different nucleotide composition from the core despite a long history of coexistence.
Figures
References
-
- Alsmark CM, Frank AC, Karlberg EO, Legault B-A, Ardell DH, Canback B, Eriksson A-S, Naslund AK, Handley SA, Huvet M, et al. The louse-borne human pathogen Bartonella quintana is a genomic derivative of the zoonotic agent Bartonella henselae. Proc Natl Acad Sci USA. 2004;101:9716–9721. - PMC - PubMed
-
- Jumas-Bilak E, Michaux-Charachon S, Bourg G, O'Callaghan D, Ramuz M. Differences in chromosome number and genome rearrangements in the genus Brucella. Mol Microbiol. 1998;27:99–106. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
