

CA150 expression delays striatal cell death in overexpression and knock-in conditions for mutant huntingtin neurotoxicity
- PMID: 16641246
- PMCID: PMC6674076
- DOI: 10.1523/JNEUROSCI.5409-05.2006
CA150 expression delays striatal cell death in overexpression and knock-in conditions for mutant huntingtin neurotoxicity
Abstract
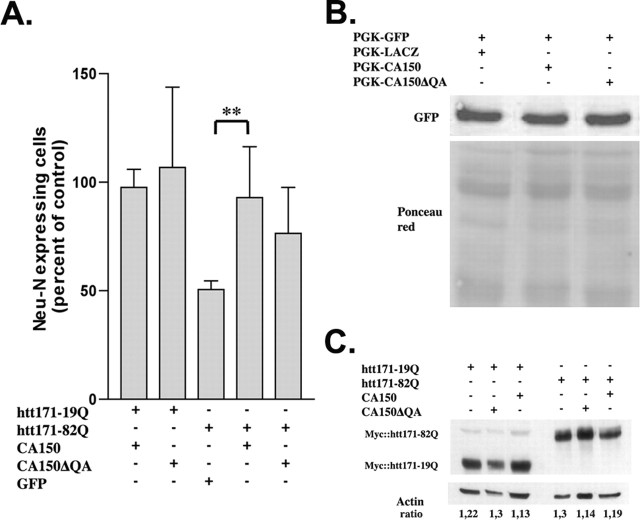
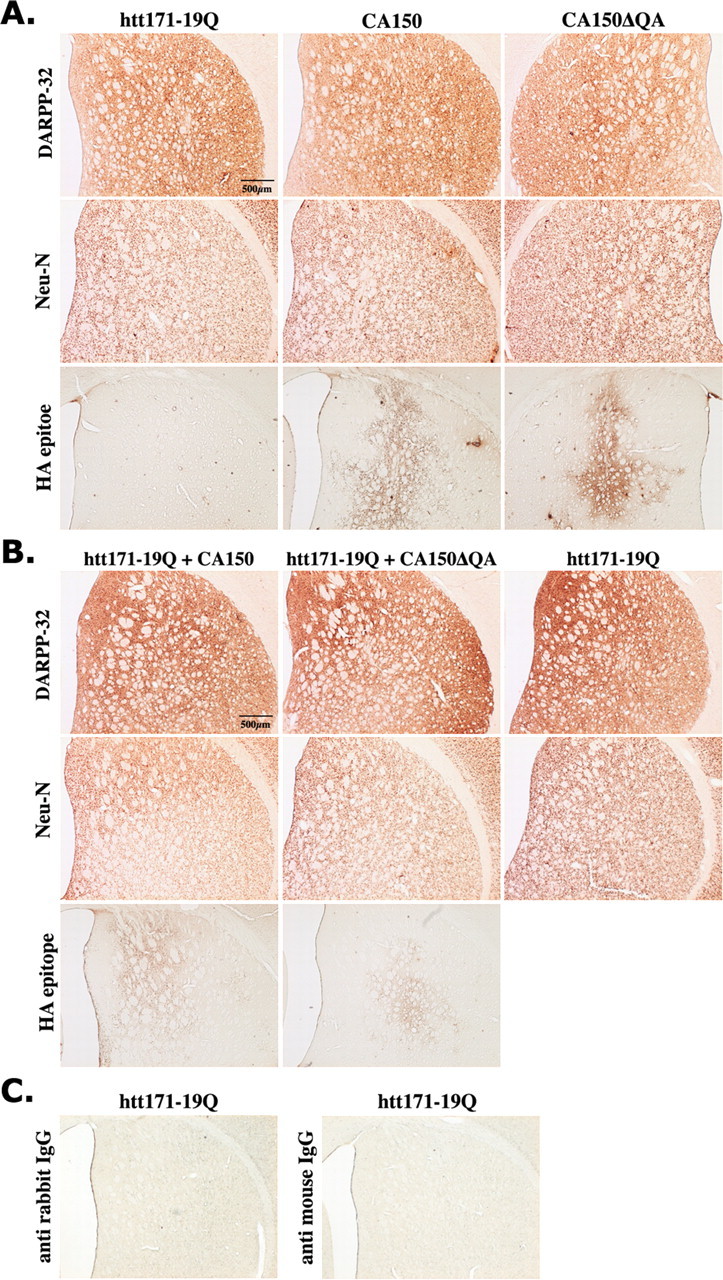
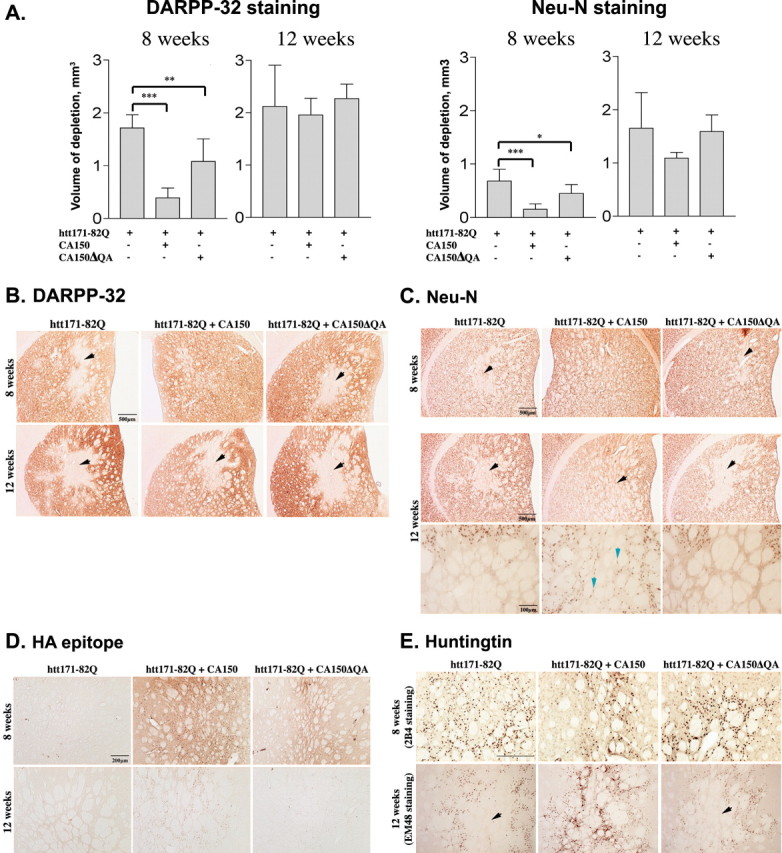
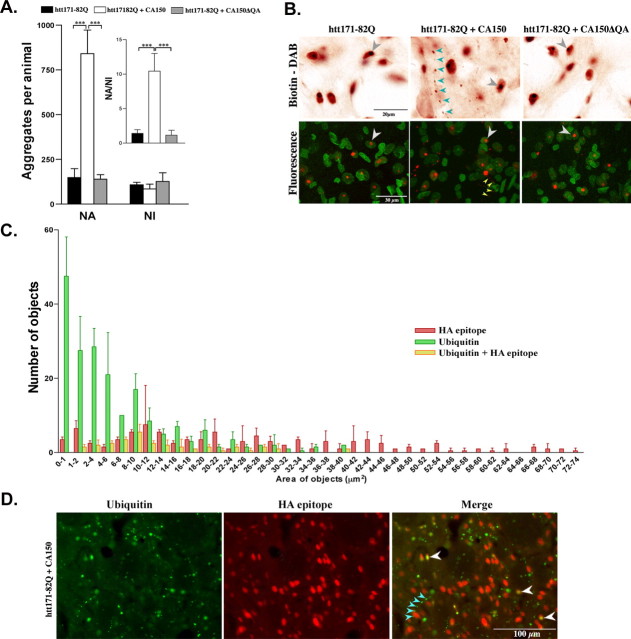
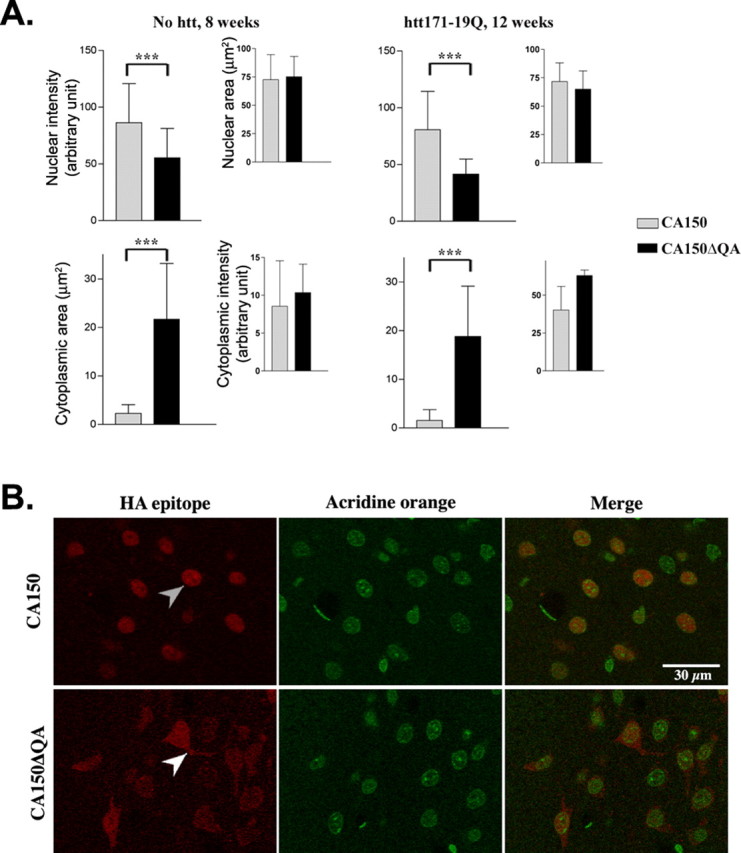
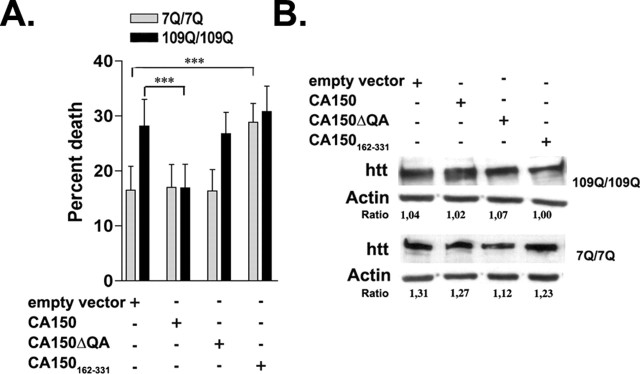
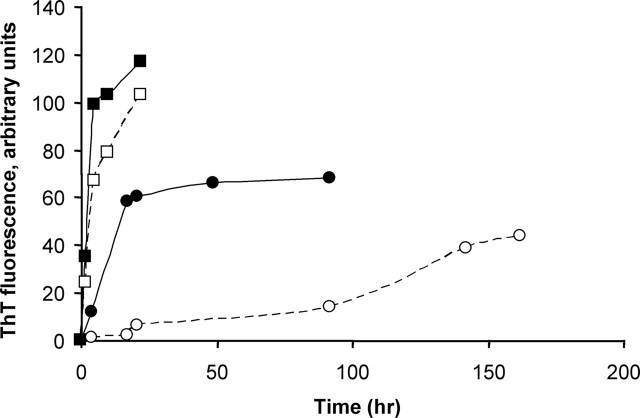
Transcriptional dysregulation caused by expanded polyglutamines (polyGlns) in huntingtin (htt) may be central to cell-autonomous mechanisms for neuronal cell death in Huntington's disease (HD) pathogenesis. We hypothesized that these mechanisms may involve the dysfunction of the transcriptional regulator CA150, a putative modifier of onset age in HD, because it binds to htt and accumulates in an HD grade-dependent manner in striatal and cortical neurons. Consistently, we report herein that CA150 expression rescues striatal cell death in lentiviral overexpression (rats) and knock-in (mouse cells) conditions for mutant htt neurotoxicity. In both systems, rescue was dependent on the (Gln-Ala)38 repeat normally found in CA150. We excluded the possibility that rescue may be caused by the (Gln-Ala)38 repeat interacting with polyGlns and, by doing so, blocking mutant htt toxicity. In contrast, we found the (Gln-Ala)38 repeat is required for the nuclear restriction of exogenous CA150, suggesting that rescue requires nuclear CA150. Additionally, we found the (Gln-Ala)38 repeat was dispensable for CA150 transcriptional repression ability, suggesting further that CA150 localization is critical to rescue. Finally, rescue was associated with increased neuritic aggregation, with no reduction of nuclear inclusions, suggesting the solubilization and nuclear export of mutant htt. Together, our data indicate that mutant htt may induce CA150 dysfunction in striatal neurons and suggest that the restoration of nuclear protein cooperativity may be neuroprotective.
Figures
Similar articles
-
The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in Huntington's disease pathogenesis.Proc Natl Acad Sci U S A. 2001 Feb 13;98(4):1811-6. doi: 10.1073/pnas.98.4.1811. Epub 2001 Jan 30. Proc Natl Acad Sci U S A. 2001. PMID: 11172033 Free PMC article.
-
Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington's disease.Neurobiol Dis. 2008 Jul;31(1):80-8. doi: 10.1016/j.nbd.2008.03.010. Epub 2008 Apr 16. Neurobiol Dis. 2008. PMID: 18502655 Free PMC article.
-
Adenovirus vector-based in vitro neuronal cell model for Huntington's disease with human disease-like differential aggregation and degeneration.J Gene Med. 2012 Jul;14(7):468-81. doi: 10.1002/jgm.2641. J Gene Med. 2012. PMID: 22700462
-
The selective vulnerability of nerve cells in Huntington's disease.Neuropathol Appl Neurobiol. 2001 Feb;27(1):1-21. doi: 10.1046/j.0305-1846.2001.00299.x. Neuropathol Appl Neurobiol. 2001. PMID: 11298997 Review.
-
Nature and cause of mitochondrial dysfunction in Huntington's disease: focusing on huntingtin and the striatum.J Neurochem. 2010 Jul;114(1):1-12. doi: 10.1111/j.1471-4159.2010.06741.x. Epub 2010 Apr 9. J Neurochem. 2010. PMID: 20403078 Review.
Cited by
-
Differential effects of sumoylation on transcription and alternative splicing by transcription elongation regulator 1 (TCERG1).J Biol Chem. 2010 May 14;285(20):15220-15233. doi: 10.1074/jbc.M109.063750. Epub 2010 Mar 9. J Biol Chem. 2010. PMID: 20215116 Free PMC article.
-
Dopamine determines the vulnerability of striatal neurons to the N-terminal fragment of mutant huntingtin through the regulation of mitochondrial complex II.Hum Mol Genet. 2008 May 15;17(10):1446-56. doi: 10.1093/hmg/ddn033. Epub 2008 Feb 11. Hum Mol Genet. 2008. PMID: 18267960 Free PMC article.
-
Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease.PLoS One. 2008 Oct 8;3(10):e3341. doi: 10.1371/journal.pone.0003341. PLoS One. 2008. PMID: 18841197 Free PMC article.
-
The Wnt receptor Ryk reduces neuronal and cell survival capacity by repressing FOXO activity during the early phases of mutant huntingtin pathogenicity.PLoS Biol. 2014 Jun 24;12(6):e1001895. doi: 10.1371/journal.pbio.1001895. eCollection 2014 Jun. PLoS Biol. 2014. PMID: 24960609 Free PMC article.
-
Proteins Containing Expanded Polyglutamine Tracts and Neurodegenerative Disease.Biochemistry. 2017 Mar 7;56(9):1199-1217. doi: 10.1021/acs.biochem.6b00936. Epub 2017 Feb 21. Biochemistry. 2017. PMID: 28170216 Free PMC article. Review.
References
-
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810. - PubMed
-
- Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY (2005). Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet 14:679–691. - PubMed
-
- Carty SM, Greenleaf AL (2002). Hyperphosphorylated C-terminal repeat domain-associating proteins in the nuclear proteome link transcription to DNA/chromatin modification and RNA processing. Mol Cell Proteomics 1:598–610. - PubMed
-
- Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S (2001). Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci 24:182–188. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases