

Sodium antimony gluconate induces generation of reactive oxygen species and nitric oxide via phosphoinositide 3-kinase and mitogen-activated protein kinase activation in Leishmania donovani-infected macrophages
- PMID: 16641451
- PMCID: PMC1472228
- DOI: 10.1128/AAC.50.5.1788-1797.2006
Sodium antimony gluconate induces generation of reactive oxygen species and nitric oxide via phosphoinositide 3-kinase and mitogen-activated protein kinase activation in Leishmania donovani-infected macrophages
Abstract
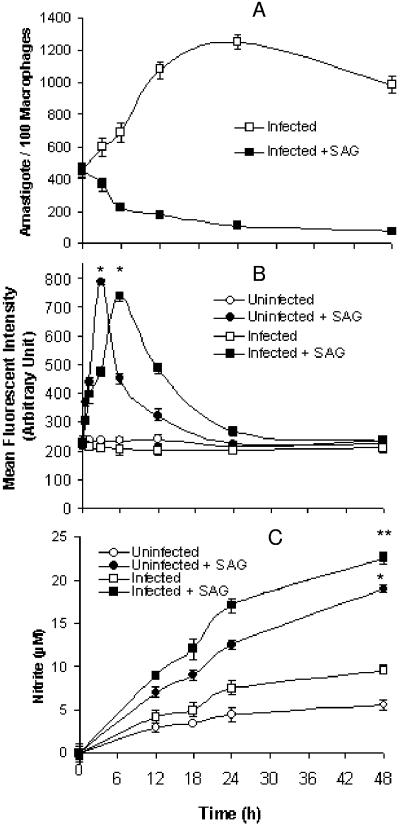
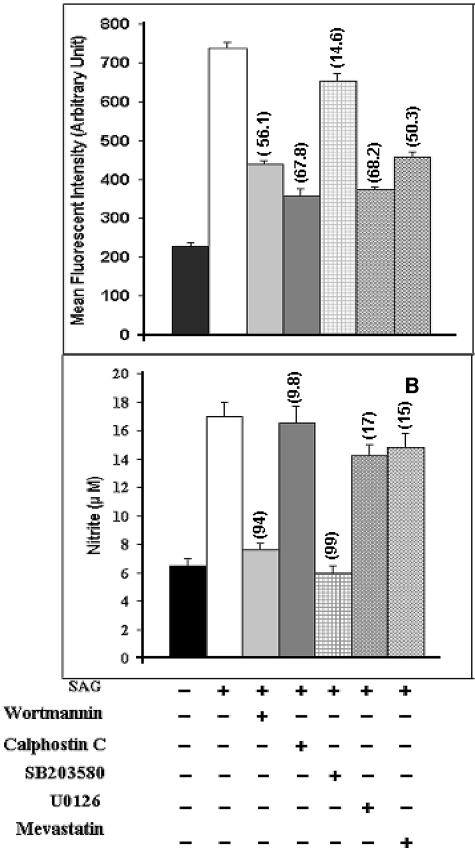
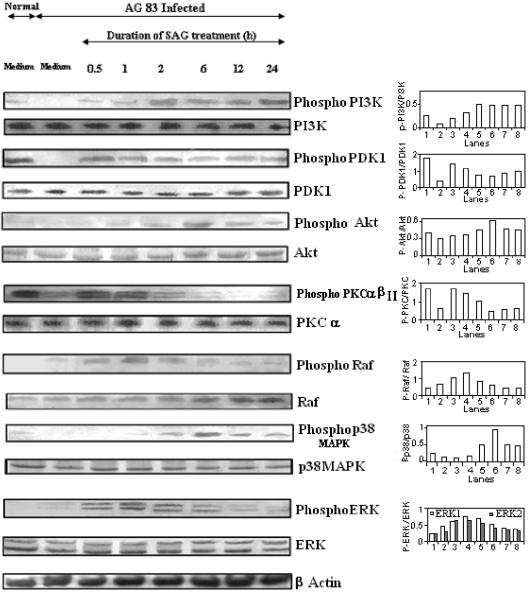
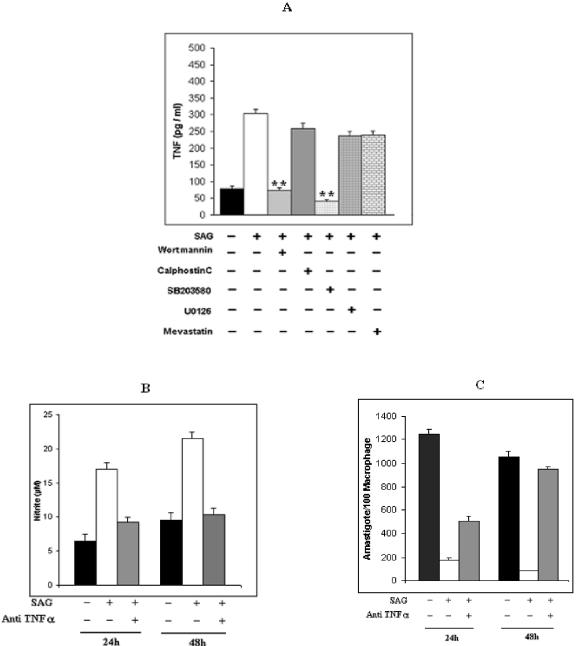
Pentavalent antimony complexes, such as sodium stibogluconate and sodium antimony gluconate (SAG), are still the first choice for chemotherapy against various forms of leishmaniasis, including visceral leishmaniasis, or kala-azar. Although the requirement of a somewhat functional immune system for the antileishmanial action of antimony was reported previously, the cellular and molecular mechanism of action of SAG was not clear. Herein, we show that SAG induces extracellular signal-regulated kinase 1 (ERK-1) and ERK-2 phosphorylation through phosphoinositide 3-kinase (PI3K), protein kinase C, and Ras activation and p38 mitogen-activated protein kinase (MAPK) phosphorylation through PI3K and Akt activation. ERK-1 and ERK-2 activation results in an increase in the production of reactive oxygen species (ROS) 3 to 6 h after SAG treatment, while p38 MAPK activation and subsequent tumor necrosis factor alpha release result in the production of nitric oxide (NO) 24 h after SAG treatment. Thus, this study has provided the first evidence that SAG treatment induces activation of some important components of the intracellular signaling pathway, which results in an early wave of ROS-dependent parasite killing and a stronger late wave of NO-dependent parasite killing. This opens up the possibility of this metalloid chelate being used in the treatment of various diseases either alone or in combination with other drugs and vaccines.
Figures


References
-
- Alexander, J., K. C. Carter, N. Al-Fasi, A. Satoskar, and F. Brombacher. 2000. Endogenous IL-4 is necessary for effective drug therapy against visceral leishmaniasis. Eur. J. Immunol. 30:2935-2943. - PubMed
-
- Amer, A. O., and M. S. Swanson. 2002. Phagosome of one's own: a microbial guide to life in the macrophage. Curr. Opin. Microbiol. 5:56-61. - PubMed
-
- Awasthi, A., R. Mathur, A. Khan, B. N. Joshi, N. Jain, S. Sawant, R. Boppana, D. Mitra, and B. Saha. 2003. CD40 signaling is impaired in L. major-infected macrophages and is rescued by a p38MAPK activator establishing a host-protective memory T cell response. J. Exp. Med. 197:1037-1043. - PMC - PubMed
-
- Berman, J. D., and D. J. Wyler. 1980. An in vitro model for investigation of chemotherapeutic agents in leishmaniasis. J. Infect. Dis. 142:83-86. - PubMed
-
- Blanchette, J., N. Racette, R. Faure, K. A. Siminovitch, and M. Olivier. 1999. Leishmania-induced increases in activation of macrophage SHP-1 tyrosine phosphatase are associated with impaired IFN-gamma-triggered JAK2 activation. Eur. J. Immunol. 29:3737-3744. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
