

Iterative gene prediction and pseudogene removal improves genome annotation
- PMID: 16651666
- PMCID: PMC1457044
- DOI: 10.1101/gr.4766206
Iterative gene prediction and pseudogene removal improves genome annotation
Abstract
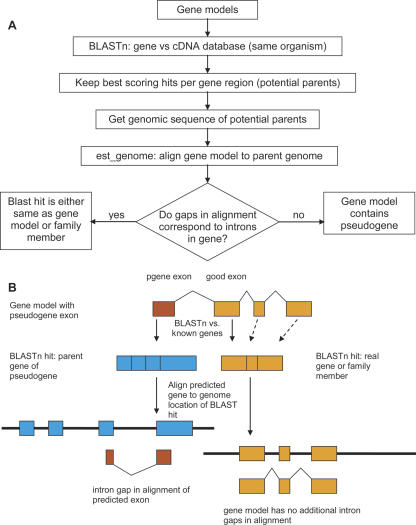
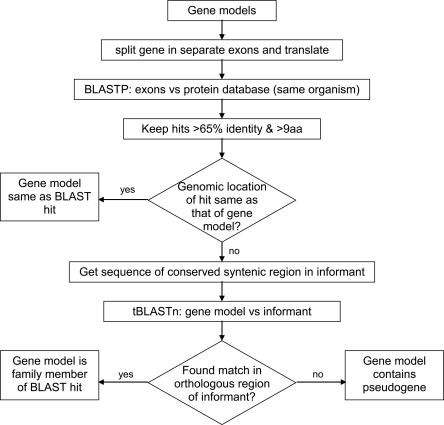
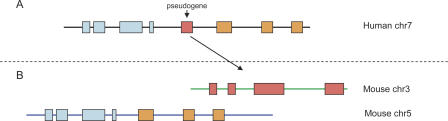
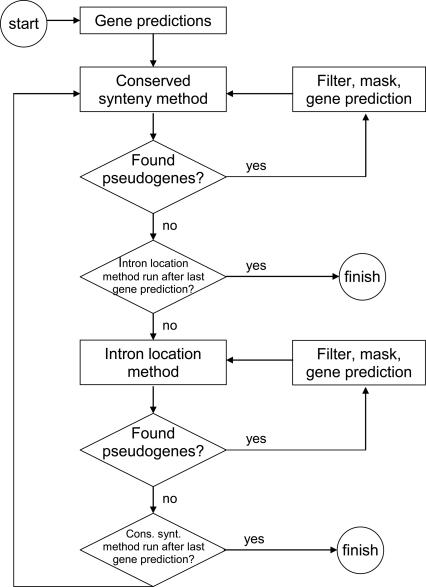
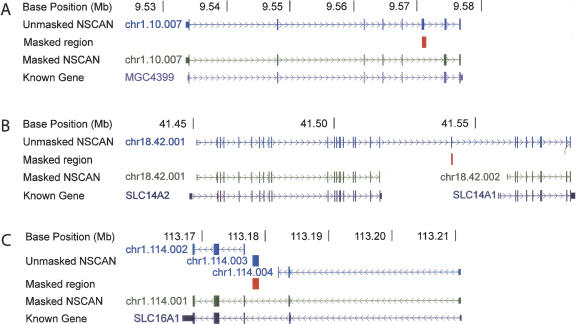
Correct gene prediction is impaired by the presence of processed pseudogenes: nonfunctional, intronless copies of real genes found elsewhere in the genome. Gene prediction programs frequently mistake processed pseudogenes for real genes or exons, leading to biologically irrelevant gene predictions. While methods exist to identify processed pseudogenes in genomes, no attempt has been made to integrate pseudogene removal with gene prediction, or even to provide a freestanding tool that identifies such erroneous gene predictions. We have created PPFINDER (for Processed Pseudogene finder), a program that integrates several methods of processed pseudogene finding in mammalian gene annotations. We used PPFINDER to remove pseudogenes from N-SCAN gene predictions, and show that gene prediction improves substantially when gene prediction and pseudogene masking are interleaved. In addition, we used PPFINDER with gene predictions as a parent database, eliminating the need for libraries of known genes. This allows us to run the gene prediction/PPFINDER procedure on newly sequenced genomes for which few genes are known.
Figures
References
-
- Ashurst J.L., Chen C.K., Gilbert J.G., Jekosch K., Keenan S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Chen C.K., Gilbert J.G., Jekosch K., Keenan S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Gilbert J.G., Jekosch K., Keenan S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Jekosch K., Keenan S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Keenan S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Searle S.M., Stalker J., Storey R., Trevanion S., Stalker J., Storey R., Trevanion S., Storey R., Trevanion S., Trevanion S., et al. The Vertebrate Genome Annotation (Vega) database. Nucleic Acids Res. 2005;33:D459–D465. - PMC - PubMed
-
- Blanco E., Parra G., Guigo R., Parra G., Guigo R., Guigo R.2003. Using geneid to identify genes. In Current protocols in bioinformatics (ed. D.B. Davison) pp. Unit 4.3. John Wiley & Sons Inc. New York
-
- Burge C., Karlin S., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases