

IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6
- PMID: 16670770
- PMCID: PMC1451201
- DOI: 10.1172/JCI21404
IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6
Abstract
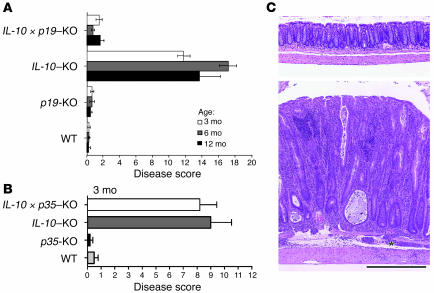
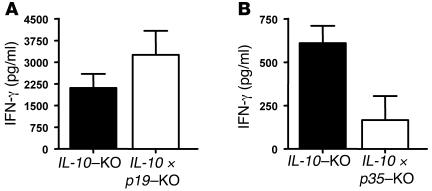
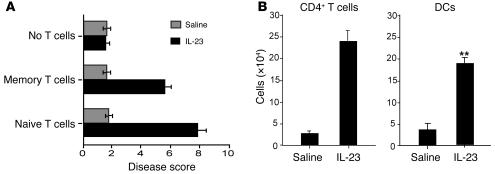
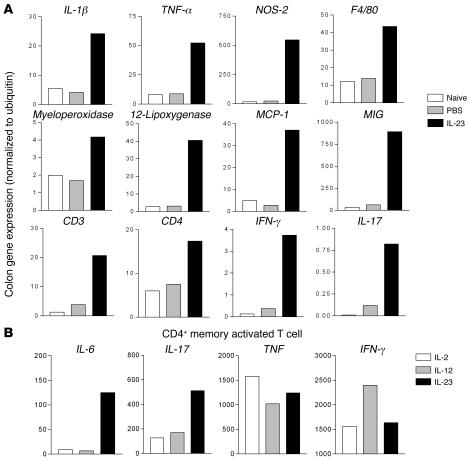
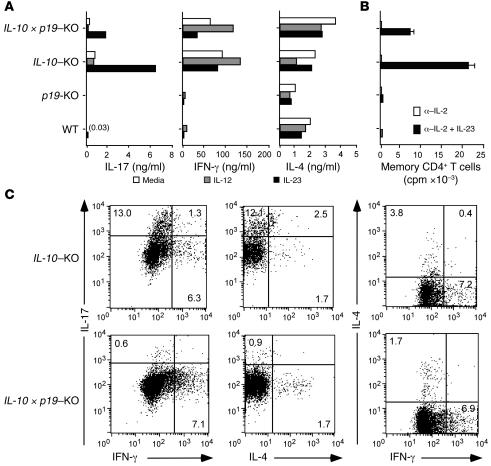
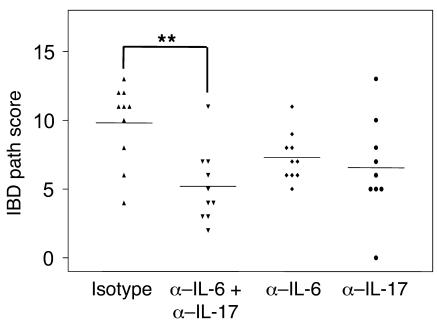
Uncontrolled mucosal immunity in the gastrointestinal tract of humans results in chronic inflammatory bowel disease (IBD), such as Crohn disease and ulcerative colitis. In early clinical trials as well as in animal models, IL-12 has been implicated as a major mediator of these diseases based on the ability of anti-p40 mAb treatment to reverse intestinal inflammation. The cytokine IL-23 shares the same p40 subunit with IL-12, and the anti-p40 mAbs used in human and mouse IBD studies neutralized the activities of both IL-12 and IL-23. IL-10-deficient mice spontaneously develop enterocolitis. To determine how IL-23 contributes to intestinal inflammation, we studied the disease susceptibility in the absence of either IL-23 or IL-12 in this model, as well as the ability of recombinant IL-23 to exacerbate IBD induced by T cell transfer. Our study shows that in these models, IL-23 is essential for manifestation of chronic intestinal inflammation, whereas IL-12 is not. A critical target of IL-23 is a unique subset of tissue-homing memory T cells, which are specifically activated by IL-23 to produce the proinflammatory mediators IL-17 and IL-6. This pathway may be responsible for chronic intestinal inflammation as well as other chronic autoimmune inflammatory diseases.
Figures
Comment in
- J Clin Invest. 116:1218.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
