

Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis
- PMID: 16670771
- PMCID: PMC1450386
- DOI: 10.1172/JCI25308
Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis
Abstract
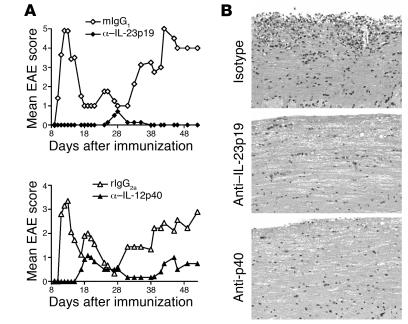
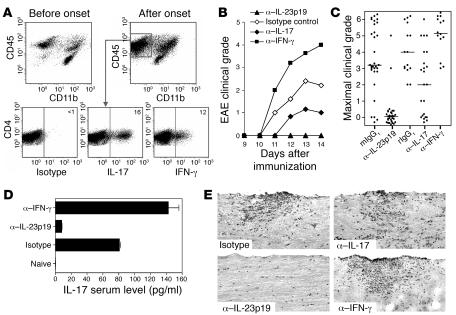
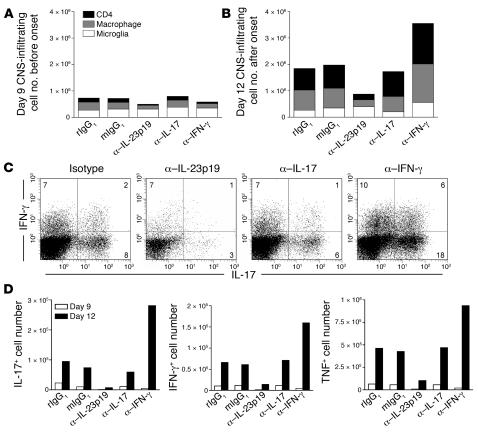
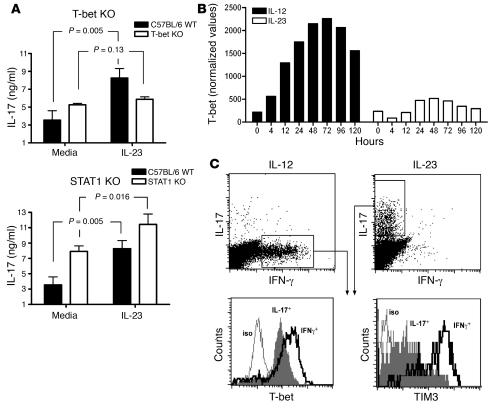
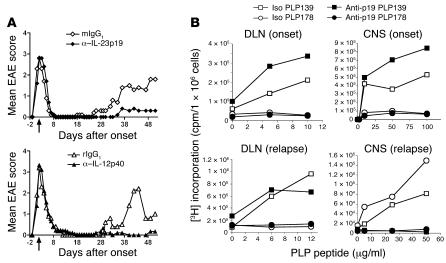
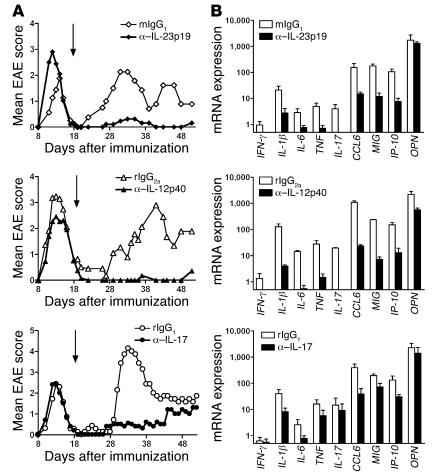
IL-23 is a member of the IL-12 cytokine family that drives a highly pathogenic T cell population involved in the initiation of autoimmune diseases. We have shown that IL-23-dependent, pathogenic T cells produced IL-17 A, IL-17 F, IL-6, and TNF but not IFN-gamma or IL-4. We now show that T-bet and STAT1 transcription factors are not required for the initial production of IL-17. However, optimal IL-17 production in response to IL-23 stimulation appears to require the presence of T-bet. To explore the clinical efficacy of targeting the IL-23 immune pathway, we generated anti-IL-23p19-specific antibodies and tested to determine whether blocking IL-23 function can inhibit EAE, a preclinical animal model of human multiple sclerosis. Anti-IL-23p19 treatment reduced the serum level of IL-17 as well as CNS expression of IFN-gamma, IP-10, IL-17, IL-6, and TNF mRNA. In addition, therapeutic treatment with anti-IL-23p19 during active disease inhibited proteolipid protein (PLP) epitope spreading and prevented subsequent disease relapse. Thus, therapeutic targeting of IL-23 effectively inhibited multiple inflammatory pathways that are critical for driving CNS autoimmune inflammation.
Figures
References
-
- Martino G., Hartung H.P. Immunopathogenesis of multiple sclerosis: the role of T cells. Curr. Opin. Neurol. 1999;12:309–321. - PubMed
-
- Behi M.E., et al. New insights into cell responses involved in experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol. Lett. 2005;96:11–26. - PubMed
-
- O’Garra A., Arai N. The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol. 2000;10:542–550. - PubMed
-
- Shtrichman R., Sanuel C.E. The role of gamma interferon in antimicrobial immunity. Curr. Opin. Microbiol. 2001;4:251–259. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
