

doi: 10.1084/jem.20051556.
Epub 2006 May 8.
The c-FLIP-NH2 terminus (p22-FLIP) induces NF-kappaB activation
Affiliations
- PMID: 16682493
- PMCID: PMC2121210
- DOI: 10.1084/jem.20051556
Item in Clipboard
The c-FLIP-NH2 terminus (p22-FLIP) induces NF-kappaB activation
J Exp Med.
.
Abstract
c-FLIP proteins (isoforms: c-FLIP(L), c-FLIP(S), and c-FLIP(R)) play an essential role in the regulation of death receptor-induced apoptosis. Here, we demonstrate that the cytoplasmic NH2-terminal procaspase-8 cleavage product of c-FLIP (p22-FLIP) found in nonapoptotic malignant cells, primary T and B cells, and mature dendritic cells (DCs) strongly induces nuclear factor kappaB (NF-kappaB) activity by interacting with the IkappaB kinase (IKK) complex via the IKKgamma subunit. Thus, in addition to inhibiting apoptosis by binding to the death-inducing signaling complex, our data demonstrate a novel mechanism by which c-FLIP controls NF-kappaB activation and life/death decisions in lymphocytes and DCs.
Figures
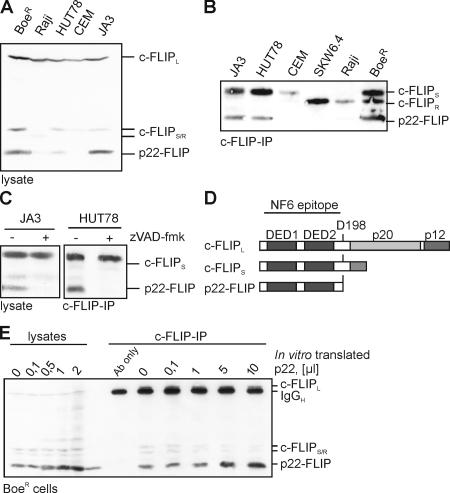
Caspase-dependent presence of p22-FLIP in tumor cell lines. (A) Total cellular lysates of the indicated T and B cell lines were subjected to 12% SDS-PAGE and Western blot analysis using the anti-FLIP mAb NF6. The positions of c-FLIPL, c-FLIPS/R, and p22-FLIP are indicated. (B) Western blot analysis of c-FLIP proteins after immunoprecipitation from various cell lines (5 × 107 cells each) using anti-FLIP mAb NF6. Positions of c-FLIPS/R and p22-FLIP are indicated. (C) HUT78 and Jurkat A3 cells were preincubated with or without 20 μM zVAD-fmk for 30 min. Analysis of c-FLIP proteins by Western blot was performed as in A. (D) Schematic representation of c-FLIP proteins. DEDs are depicted in black. The cleavage site for generation of p22-FLIP (D198) is shown. The epitope for anti–c-FLIP mAb NF6 is indicated. (E) The NH2 terminus of c-FLIP encoding the amino acids 1–198 was in vitro translated, [35S] labeled, and added in the indicated amounts to the lysates of BoeR cells as well as to immunoprecipitates of c-FLIP from 5 × 107 BoeR cells.
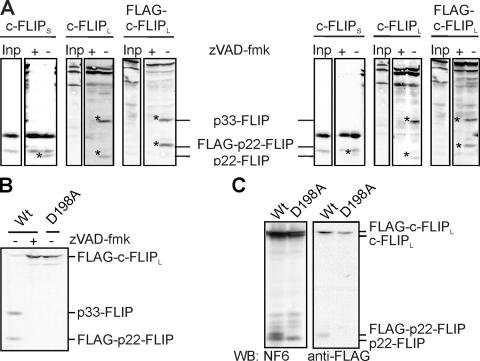
Identification of the p22-FLIP cleavage site. (A) In vitro–translated [35S]-labeled c-FLIPS, c-FLIPL, or FLAG-c-FLIPL was added to the lysates of HUT78 (left) and Jurkat A3 cells (right) and incubated overnight at 4°C in presence or absence of 50 μM zVAD-fmk. Reactions were separated on 12% SDS-PAGE gels, blotted, and subjected to autoradiography. (B) In vitro–translated [35S]-labeled WT-FLAG-c-FLIPL or D198A-FLAG-c-FLIPL was added to the lysates of HUT78 cells and incubated overnight at 4°C in the presence or absence of 50 μM zVAD-fmk and visualized as in A. (C) BoeR cells were transfected with WT-FLAG-c-FLIPL or D198A-FLAG-c-FLIPL and incubated with or without 20 μM zVAD-fmk, and c-FLIP proteins were analyzed by Western blot.
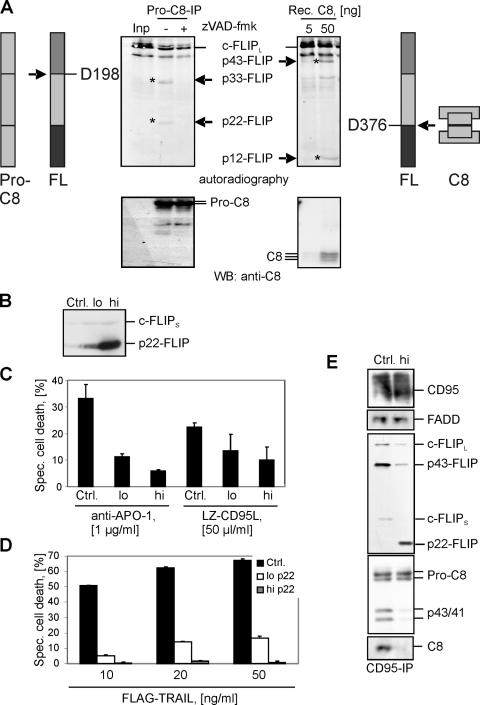
p22-FLIP is generated by procaspase-8 and inhibits death receptor–induced apoptosis. (A) Procaspase-8 was immunoprecipitated from HUT78 cells using anti–caspase-8 mAb C15 and then incubated for 1 h at 37°C together with in vitro–translated [35S]-labeled c-FLIPL in the presence or absence of zVAD-fmk. c-FLIP processing was analyzed by autoradiography (top left). c-FLIP cleavage products p22 and p33 are indicated. Afterward, the same membrane was subjected to Western blot analysis using anti–caspase-8 mAb C15 (bottom left). [35S]-labeled c-FLIPL was incubated with the indicated concentrations of recombinant caspase-8 for 1 h at 37°C. c-FLIP processing was analyzed byautoradiography (top right). c-FLIP cleavage products p12 and p43 are indicated. Afterward, the same membrane was subjected to Western blot analysis using anti–caspase-8 mAb C15 (bottom right). (B) Analysis of p22-FLIP expression in BJAB cell lines stably overexpressing high or low amounts of p22-FLIP (p22-FLIPhigh or p22-FLIPlow, respectively). Endogenous expression of c-FLIPS is used as a loading control. (C) p22-FLIPhigh, p22-FLIPlow, and vector-transfected BJABs (Ctrl.) were stimulated with 1 μg/ml anti–APO-1 antibodies or 50 μl/ml LZ-CD95L for 16 h. Specific cell death was calculated as described in Materials and methods. (D) p22-FLIPhigh, p22-FLIPlow, and vector-transfected BJABs (Ctrl.) were stimulated with the indicated concentrations of FLAG-TRAIL for 16 h. (E) CD95 DISCs were immunoprecipitated from 5 × 107 cells of p22-FLIPhigh and vector-transfected BJABs (Ctrl.) and analyzed by Western blot with anti–caspase-8 mAb C15, anti-FLIP mAb NF6, anti-CD95 polyclonal antibody C20, and anti-FADD mAb.
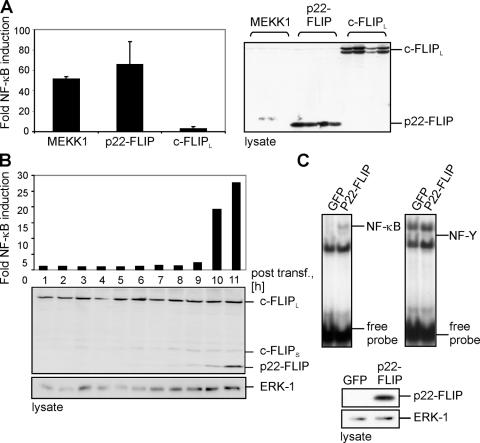
p22-FLIP is a strong inducer of NF-κB. (A) 293T cells were cotransfected with MEKK1, p22-FLIP, c-FLIPL, and luciferase reporter plasmid. GFP transfections were performed to control the transfection efficiency. Western blot analysis using anti-FLIP mAb NF6 was performed to control equal protein expression (right). (B) 293T cells were cotransfected with p22-FLIP and the luciferase reporter plasmid. After the indicated periods of time, cells were lysed and NF-κB luciferase activity was determined (top). Western blot analysis using anti-FLIP mAb NF6 was performed to determine the expression level of p22-FLIP. (C) Nuclear extracts, which were prepared from 293T cells transfected with p22-FLIP or GFP, were subjected to EMSAs using 32P-labeled oligonucleotides containing an NF-κB (left) or an NF-Y (right) binding site. p22-FLIP expression was verified by Western blot.
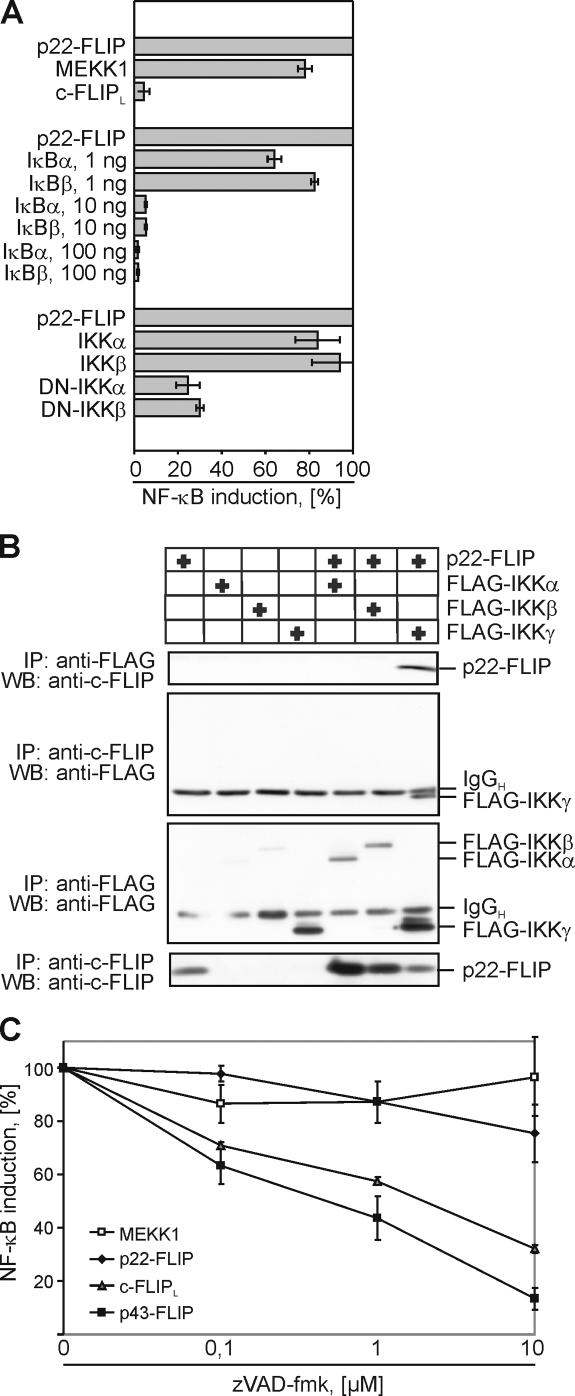
p22-FLIP induces NF-κB by direct interaction with the IKK complex. (A) 293T cells were cotransfected with luciferase reporter plasmid and either MEKK1, p22-FLIP, or c-FLIPL (top part of the diagram). 293T cells were cotransfected with p22-FLIP, the luciferase reporter plasmid, and any one of the constructs IκBα, IκBβ, WT-IKKα, WT-IKKβ, mutated IKKα, or IKKβ (bottom part of the diagram). Transfection efficiency was examined using GFP transfections. NF-κB luciferase activity was determined as described in Materials and methods. (B) FLAG or FLIP immunoprecipitations were performed from 293T cells that were transfected with p22-FLIP and any one of the constructs FLAG-IKKα, FLAG-IKKβ, or FLAG-IKKγ. Immunoprecipitated products were subjected to 12% SDS-PAGE gels and analyzed by Western blot using anti-FLIP mAb NF6 and anti-FLAG mAb. (C) 293T cells were cotransfected with MEKK1, p22-FLIP, c-FLIPL, p43-FLIP, and the luciferase reporter plasmid. Transfected cells were incubated for 16 h in the presence of the indicated concentrations of zVAD-fmk and lysed, and NF-κB luciferase activity was determined.
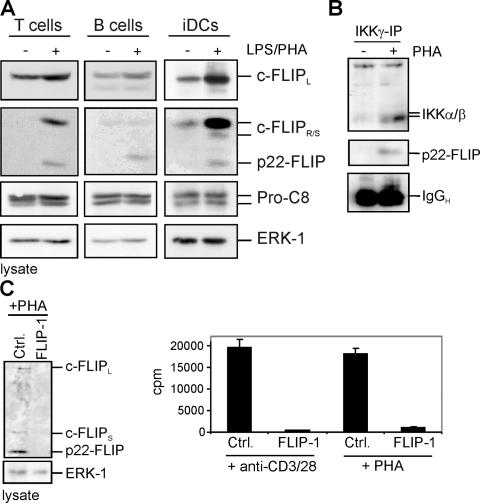
p22-FLIP is a key mediator of NF-κB induction. (A) Primary human T and B cells were stimulated with 1 μg/ml PHA. Primary immature iDCs were stimulated with 500 ng/ml LPS. Western blot analysis was performed using the anti-FLIP mAb NF6, the anti–caspase-8 mAb C15, and the anti-ERK1 mAb. (B) 107 primary human T cells were stimulated with 1 μg/ml PHA for 12 h and lysed, and anti-IKKγ immunoprecipitation was performed. Western blot analysis was performed using anti-IKKα/β antibody and anti-FLIP mAb NF6. (C) Primary human T cells were transiently transfected with double-stranded siRNA oligonucleotides comprising a FLIP-specific sequence (FLIP-1) or a nonspecific sequence (Ctrl.). 48 h after transfection, cells were stimulated with 1 μg/ml PHA for 3 d (right) or 0.5 μg/ml anti-CD3/28 for 24 h (left) or 3 d (right). After incubation, cells were lysed and Western blot analysis was performed using the anti-FLIP mAb NF6 (left), or the proliferation was measured after incorporation of tritiated thymidine ([3H]TdR) during the last 18 h.
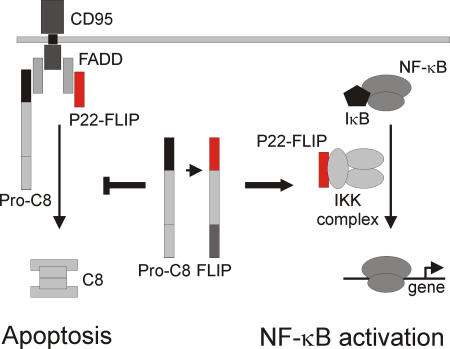
The dual function of p22-FLIP in the cell. p22-FLIP induces NF-κB by interacting with IKKγ in the IKK complex (right side). In addition, p22-FLIP can block death receptor–mediated apoptosis by binding to the DISC via DED interactions and inhibiting procaspase-8 activation (left side).
References
-
- Golks, A., D. Brenner, C. Fritsch, P.H. Krammer, and I.N. Lavrik. 2005. c-FLIPR, a new regulator of death receptor-induced apoptosis. J. Biol. Chem. 280:14507–14513. - PubMed
-
- Krueger, A., I. Schmitz, S. Baumann, P.H. Krammer, and S. Kirchhoff. 2001. Cellular flice-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the cd95 death-inducing signaling complex. J. Biol. Chem. 276:20633–20640. - PubMed
-
- Scaffidi, C., I. Schmitz, P.H. Krammer, and M.E. Peter. 1999. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 274:1541–1548. - PubMed
-
- Thome, M., P. Schneider, K. Hofmann, H. Fickenscher, E. Meinl, F. Neipel, C. Mattmann, K. Burns, J.L. Bodmer, M. Schröter, et al. 1997. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 386:517–521. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
