

A simple physical model predicts small exon length variations
- PMID: 16683028
- PMCID: PMC1449888
- DOI: 10.1371/journal.pgen.0020045
A simple physical model predicts small exon length variations
Abstract
One of the most common splice variations are small exon length variations caused by the use of alternative donor or acceptor splice sites that are in very close proximity on the pre-mRNA. Among these, three-nucleotide variations at so-called NAGNAG tandem acceptor sites have recently attracted considerable attention, and it has been suggested that these variations are regulated and serve to fine-tune protein forms by the addition or removal of a single amino acid. In this paper we first show that in-frame exon length variations are generally overrepresented and that this overrepresentation can be quantitatively explained by the effect of nonsense-mediated decay. Our analysis allows us to estimate that about 50% of frame-shifted coding transcripts are targeted by nonsense-mediated decay. Second, we show that a simple physical model that assumes that the splicing machinery stochastically binds to nearby splice sites in proportion to the affinities of the sites correctly predicts the relative abundances of different small length variations at both boundaries. Finally, using the same simple physical model, we show that for NAGNAG sites, the difference in affinities of the neighboring sites for the splicing machinery accurately predicts whether splicing will occur only at the first site, splicing will occur only at the second site, or three-nucleotide splice variants are likely to occur. Our analysis thus suggests that small exon length variations are the result of stochastic binding of the spliceosome at neighboring splice sites. Small exon length variations occur when there are nearby alternative splice sites that have similar affinity for the splicing machinery.
Conflict of interest statement
Competing interests. The authors have declared that no competing interests exist.
Figures
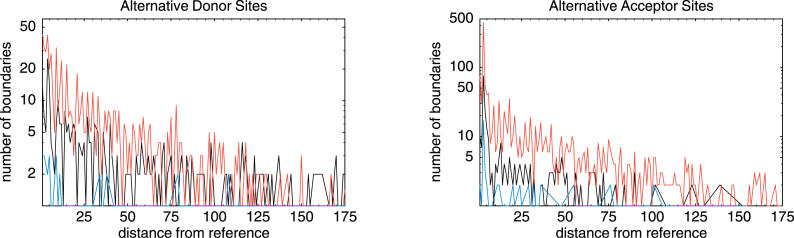
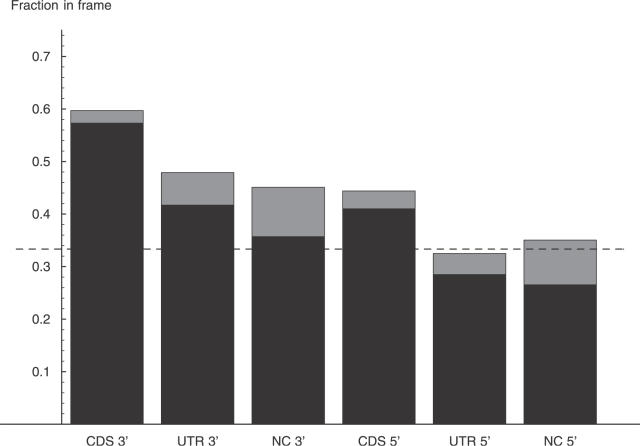
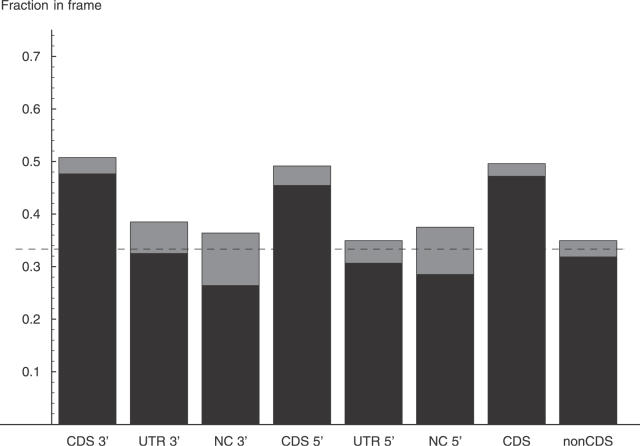
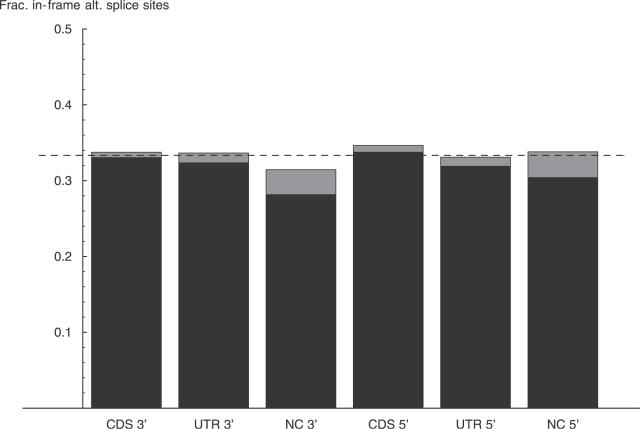
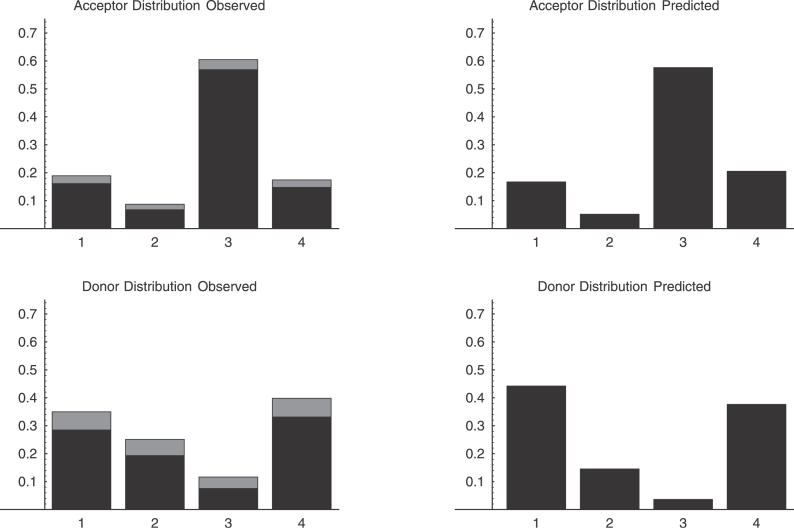

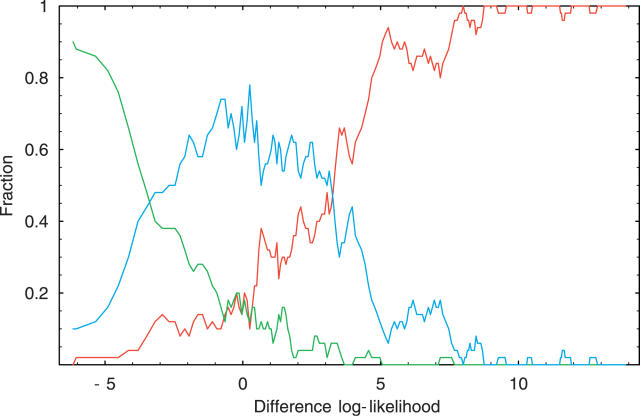
Comment in
-
Alternative splicing at NAGNAG acceptors: simply noise or noise and more?PLoS Genet. 2006 Nov 24;2(11):e207; author reply e208. doi: 10.1371/journal.pgen.0020207. PLoS Genet. 2006. PMID: 17121470 Free PMC article. No abstract available.
References
-
- Lander E, Linton L, Birren B, Nusbaum C, Zody M, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
-
- Venter J, Adams M, Myers E, Li P, Mural R, et al. The sequence of the human genome. Science. 2001;291:1304–1351. - PubMed
-
- Waterston R, Lindblad-Toh K, Birney E, Rogers J, Abril J, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. - PubMed
-
- Kawai J, Shinagawa A, Shibata K, Yoshino M, Itoh M, et al. Functional annotation of a full−length mouse cDNA collection. Nature. 2001;409:685–690. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
