

Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome
- PMID: 16684884
- PMCID: PMC1472513
- DOI: 10.1073/pnas.0600206103
Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome
Abstract
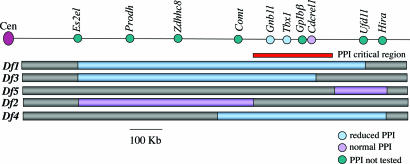
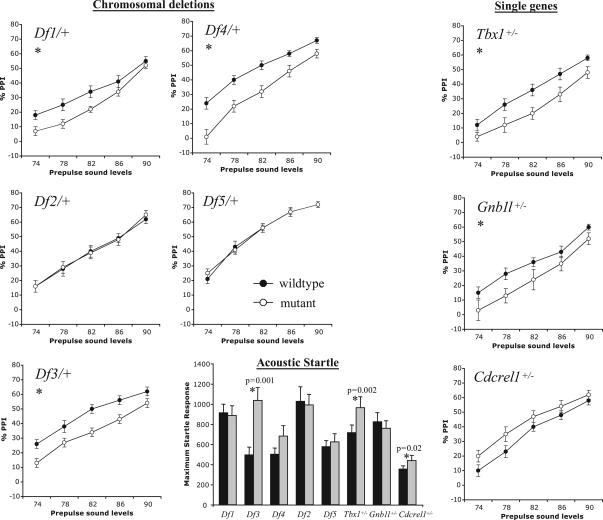
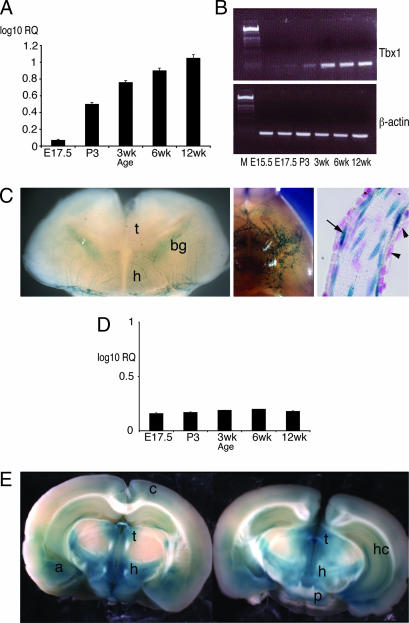
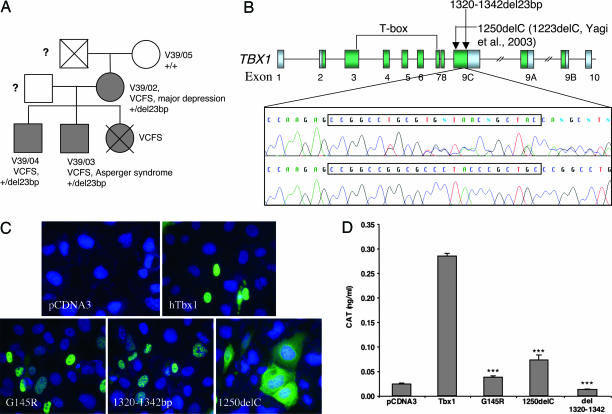
About 35% of patients with 22q11 deletion syndrome (22q11DS), which includes DiGeorge and velocardiofacial syndromes, develops psychiatric disorders, mainly schizophrenia and bipolar disorder. We previously reported that mice carrying a multigene deletion (Df1) that models 22q11DS have reduced prepulse inhibition (PPI), a behavioral abnormality and schizophrenia endophenotype. Impaired PPI is associated with several psychiatric disorders, including those that occur in 22q11DS, and recently, reduced PPI was reported in children with 22q11DS. Here, we have mapped PPI deficits in a panel of mouse mutants that carry deletions that partially overlap with Df1 and have defined a PPI critical region encompassing four genes. We then used single-gene mutants to identify the causative genes. We show that PPI deficits in Df1/+ mice are caused by haploinsufficiency of two genes, Tbx1 and Gnb1l. Mutation of either gene is sufficient to cause reduced PPI. Tbx1 is a transcription factor, the mutation of which is sufficient to cause most of the physical features of 22q11DS, but the gene had not been previously associated with the behavioral/psychiatric phenotype. A likely role for Tbx1 haploinsufficiency in psychiatric disease is further suggested by the identification of a family in which the phenotypic features of 22q11DS, including psychiatric disorders, segregate with an inactivating mutation of TBX1. One family member has Asperger syndrome, an autistic spectrum disorder that is associated with reduced PPI. Thus, Tbx1 and Gnb1l are strong candidates for psychiatric disease in 22q11DS patients and candidate susceptibility genes for psychiatric disease in the wider population.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
